

Shimin Yang1, Jianyu Chen1, Jinjian Fu1, Jiayin Huang, Ting Li, Zhenjiang Yao, and Xiaohua YeComments to Author
Author affiliations: Guangdong Pharmaceutical University, Guangzhou, China (S. Yang, J. Chen, J. Huang, T. Li, Z. Yao, X. Ye); Liuzhou Maternity and Child Health Care Hospital, Liuzhou, China (J. Fu)
Abstract
Streptococcus pneumoniae is an opportunistic pathogen that causes substantial illness and death among children worldwide. The genetic backgrounds of pneumococci that cause infection versus asymptomatic carriage vary substantially. To determine the evolutionary mechanisms of opportunistic pathogenicity, we conducted a genomic surveillance study in China. We collected 783 S. pneumoniae isolates from infected and asymptomatic children. By using a 2-stage genomewide association study process, we compared genomic differences between infection and carriage isolates to address genomic variation associated with pathogenicity. We identified 8 consensus k-mers associated with adherence, antimicrobial resistance, and immune modulation, which were unevenly distributed in the infection isolates. Classification accuracy of the best K-mer predictor for S. pneumoniae infection was good, giving a simple target for predicting pathogenic isolates. Our findings suggest that S. pneumoniae’s pathogenicity is complex and multifactorial, and we provide genetic evidence for precise targeted interventions.
Streptococcus pneumoniae is a pathogen that causes community-associated infections in young children <5 years of age 1,2. It can asymptomatically colonize the nasopharynx and upper airway in healthy children (up to 60%) and can also invade sterile sites and lead to infections from mild to life-threatening, which can result in substantial illness and death worldwide 1,3,4. Despite the widespread use of pneumococcal vaccines to immunize children, S. pneumoniae remains the leading cause of life-threatening diseases. Worldwide, the increasing disease burden of S. pneumoniae is alarming; an estimated 1 million children <5 years of age die of pneumococcal disease every year 5. All pneumococcal diseases arise from bacterial colonization, and the adaptability of the virulence characteristics enhances pneumococcal persistence in the colonization of the host respiratory tract, suggesting that nasopharyngeal carriage of S. pneumoniae plays a key role in the development and transmission of pneumococcal diseases 6. Pneumococcal disease is one of the most common infectious diseases caused by asymptomatic S. pneumoniae colonization in humans. Eliminating this opportunistic pathogenic bacterium requires knowledge of the pathogenicity-associated genetic elements that distinguish infection from carriage isolates. Previous studies have been limited to exploring virulence factors and molecular characterization of invasive S. pneumoniae isolates 7,8.
Whole-genome sequencing (WGS) has become a powerful tool for bacterial genotyping; costs have been decreasing as accessibility increases. The high-dimensional genomic data can provide unprecedented resolution for identifying subtle genomic variations. Genomewide association studies (GWAS) are increasingly used to detect novel genes and genetic elements associated with bacterial phenotypes, which may provide insight for future preventive strategies and control measures 9–12. In brief, traditional GWAS methods can be used to identify large numbers of common genetic variants, usually single-nucleotide polymorphisms (SNPs), to determine the genetic basis of bacterial phenotypes of interest. However, considering the high genomic plasticity of many species of bacteria, traditional GWAS methods can only partially identify the phenotype-associated genetic variants. To avoid the limitations of SNP-based GWAS, we used k-mers (DNA words of length k) as an alternative method, which can capture different types of variants 13,14.
To determine whether the genetic variation is unevenly enriched in S. pneumoniae infection isolates, we used multiple GWAS analyses to compare genomic differences between infection and carriage isolates. Study protocols were approved by the Ethics Committee of Guangdong Pharmaceutical University (2019–19) and the Ethics Committee of Liuzhou Maternity and Child Healthcare Hospital (2018–84). We obtained written informed consent from parents or legal guardians on behalf of the children.
Methods
Sampling
From 2015–2021, we collected clinical samples from infected children and nasal swab samples from healthy children in southern China (Guangxi and Guangdong Provinces). From hospitalized infected children, we collected 349 nonrepetitive pneumococcal isolates (e.g., blood, bronchoalveolar lavage fluid, sputum, middle ear fluid), of which 342 were noninvasive and 7 invasive. The eligibility criteria for infected children were having clinical infectious manifestations such as cough, respiratory secretions, abnormal lung sounds, dyspnea, or fever >38°C, with or without infiltrates seen on chest radiographs; having S. pneumoniae infection diagnosed by clinical doctors based on signs and symptoms; and having S. pneumoniae isolated from clinical infection sites. In terms of asymptomatic carriage isolates, we sampled 434 isolates from healthy children in kindergarten.
Whole-Genome Sequencing
We performed high-throughput genome sequencing on a Hiseq 2000 machine (Illumina, https://www.illumina.comExternal Link) to obtain paired-end 150-bp reads. We assessed the quality of the raw sequenced reads by using FastQC version 0.11.5 (https://github. com/s-Andrews/FastQCExternal Link) and trimmed for low-quality reads and adaptor regions by using Trimmomatic version 0.36 (https://github.com/usadellab/TrimmomaticExternal Link). We then assembled trimmed reads by using SPAdes version 3.6.1 (https://github.com/ablab/spadesExternal Link). We used PathogenWatch (https://pathogen.watch) to predict global pneumococcal sequencing cluster (GPSC), multilocus sequence typing (MLST), and serotyping for all genomes.
Phylogenetic Analyses
To generate the variant sites with SNPs, we mapped assembled contigs to a standard reference genome S. pneumoniae R6 by using Snippy version 4.4.5 (https://github.com/tseemann/snippyExternal Link). We used the generated core SNP alignment to construct a maximum-likelihood phylogenetic tree by using the generalized time-reversible plus gamma model and 100 bootstrap replicates with FastTree version 2.1.10 (http://www.microbesonline.org/fasttreeExternal Link). We visualized and annotated the phylogenetic tree by using ChiPlot (https://www.chiplot.online).
Counting and Annotating k-mers
We scanned all k-mers that were 9- to 100-bp long from all assembled reads by using fsm-lite (https://github.com/nvalimak/fsm-lite External Link) and filtered them to obtain 10,591,337 k-mers seen on 1%–99% of the total samples. To identify the relevant genes by using BWA-MEM (the Burrows-Wheeler Aligner with maximal exact matches alignment tool, https://github.com/lh3/bwaExternal Link), we mapped all k-mers to 10 S. pneumoniae reference genomes (CGSP14, D39, Hungary19A-6, R6, Taiwan19F-14, TIGR4, Spain23F-ST81, ATCC 49619, EF3030, and MDRSPN001) obtained from the Virulence Factor Database (http://www.mgc.ac.cn/VFsExternal Link) and previous studies. We determined gene ontology annotations by using the UniProt (https://beta.uniprot.orgexternal Link).
Multiple GWAS Analyses of Disease-Associated k-mers
To explore the genome-wide associations between genetic elements (k-mers) and S. pneumoniae disease status (infection or carriage), and thus to identify infection-associated k-mers, we used GWAS methods. Because of the high-dimensional genomic data structures, we used multiple GWAS methods: the linear mixed model (LMM; (https://github.com/mgalardini/pyseerExternal Link), phylogenetic-based approach (Scoary; https:// github.com/AdmiralenOla/ScoaryExternal Link), variable selection using random forests (VSURF; https://github.com/robingenuer/VSURFExternal Link), and least absolute shrinkage and selection operator (LASSO; https://scikit-learn.org/stable/modules/generated/sklearn.linear_model.Lasso.h tmlExternal Link) regression.
In brief, we used a 2-stage analysis process to detect the infection-associated k-mers by comprehensive GWAS analyses (Figure 1). First, we fitted a univariate LMM to initially screen infection-associated k-mers by using the Pyseer tool (version 1.3.10) 15. To correct the population structure, we used the similarity Pyseer command Pyseer, which computes a similarity kinship matrix based on the core genome SNPs. For covariates, the GWAS analysis used host age (years) and sex. Second, we used multiple methods (Scoary, LASSO, and VSURF) to minimize false-positive associations and identify consensus infection-associated k-mers by the Venn diagram. In the GWAS analyses, we used the Bonferroni correction (α/N) to control for false-positive rates resulting from multiple comparisons of 1,418,815 k-mers (adjusted p-value threshold 3.52 × 10−8). Scoary is an ultrafast software tool for GWAS analyses that uses a phylogenetic-based method to adjust population structure. The LASSO regression is suitable for high-dimensional data structures, and the coefficients of nonrelevant variables can be compressed to zero to solve the problem of model overfitting 16. We used VSURF, based on random forest (RF), to perform a 2-step feature selection on the variables 17. Initially, VSURF ranks the variables according to the importance measure by using the RF permutation-based score of importance to obtain a subset of important variables, and then it uses a stepwise forward strategy for variable introduction based on the smallest out-of-bag error. More precisely, a variable is added only if the error decrease is larger than a threshold. We ranked the importance of k-mers by the mean decrease in impurity (mean decrease Gini), which is a measure of the predictor’s contribution to the correct sample classification. We compiled associated phenotype data for all 783 isolates (Appendix Table 1) and deposited sequences in the National Center for Biotechnology Information Sequence Read Archive database (https://www.ncbi.nlm.nih.gov/ sraExternal Link; projection no. PRJNA976286). The k-mer sequences and output results files from several GWAS analyses are publicly available (https://doi.org/10.6084/m9. figshare.24466606.v3External Link).
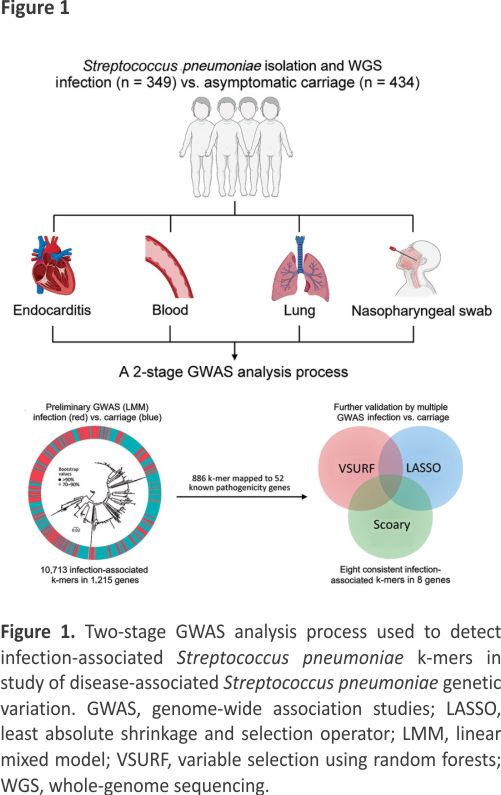
Results
Characteristics of S. pneumoniae Isolates
Of the 349 children with S. pneumoniae infection, 342 (98.0%) had noninvasive disease (264 pneumonia, 49 bronchitis, 13 otitis media, 9 upper respiratory infection, 6 nasosinusitis, and 1 corneal ulcer), and 7 (2.0%) had invasive disease (6 bacteremia and 1 endocarditis). χ2 test results indicated no differences between infection and carriage isolates concerning host sex (p = 0.359) but significant age differences (p<0.001) (Appendix Table 2).
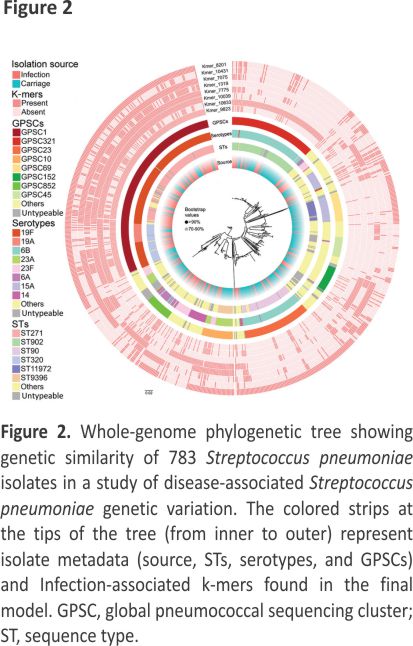
Association between Genotypes and Disease Status
The most prevalent GPSCs for infection isolates were GPSC1 (45.9%), GPSC321 (9.2%), and GPSC852 (5.4%); the predominant GPSCs for carriage isolates were GPSC321 (16.1%), GPSC1 (15.4%), and GPSC23 (15.0%). In terms of sequence types (STs), the most common genotypes for infection isolates were ST271 (29.2%), ST320 (9.5%), and ST902 (7.2%); the predominant genotypes for carriage isolates were ST902 (15.9%), ST90 (13.8%), and ST271 (8.5%). The most prevalent serotypes for infection isolates were 19F (43.0%), 6B (15.2%), and 23F (8.3%); and the predominant serotypes for carriage isolates were 6B (32.7%), 19F (13.1%), and 15A (11.1%). The results indicated potential genotype differences between infection and carriage isolates. In addition, the phylogenetic tree based on core SNPs revealed that several genotypes (GPSCs/STs/serotypes) from infection and carriage isolates clustered in the same branches (Figure 2). Moreover, we found statistically significant differences in the proportion of specific GPSCs/STs/serotypes between infection and carriage isolates (Table 1), indicating that these isolates are associated with infection.
Preliminary Screening for Infection-Associated k-mers by LMM
We identified 10,591,337 k-mers from the assemblies of 783 S. pneumoniae isolates and then filtered out low-frequency k-mers for a reduced matrix with 1,418, 815 k-mers. Using those k-mers for GWAS, we performed a univariate LMM analysis to identify 22,790 infection-associated k-mers; 10,713 k-mers were successfully mapped to 1,215 unique genes (Figure 3, panel A; Appendix Figure 1). In the initial model with 10,713 k-mers, we used the RF model to assess the prediction effect. The classification balanced accuracy based on cross-validation was 93.60% (95% CI 91.48%–95.72%) (Table 2); the area under the curve (AUC), based on the out-of-bag risk scores of the classifier, was 0.98. In the LMM analysis, the QQ plot indicated that population structure was well controlled at low p values (p<0.01) (Appendix Figure 2). Because of the considerable redundancy among the genetic elements in risk prediction, studying all k-mer combinations had little benefit; therefore, we used a simpler model with 886 k-mers successfully mapped to 52 antibiotic resistance or virulence genes (Appendix Table 3). The classification balanced accuracy was 91.28% (95% CI 89.34%– 93.22%) (Table 2); the AUC was 0.96, suggesting that the power of these 886 k-mers for predicting disease status was close to that of the model with 10,713 k-mers.
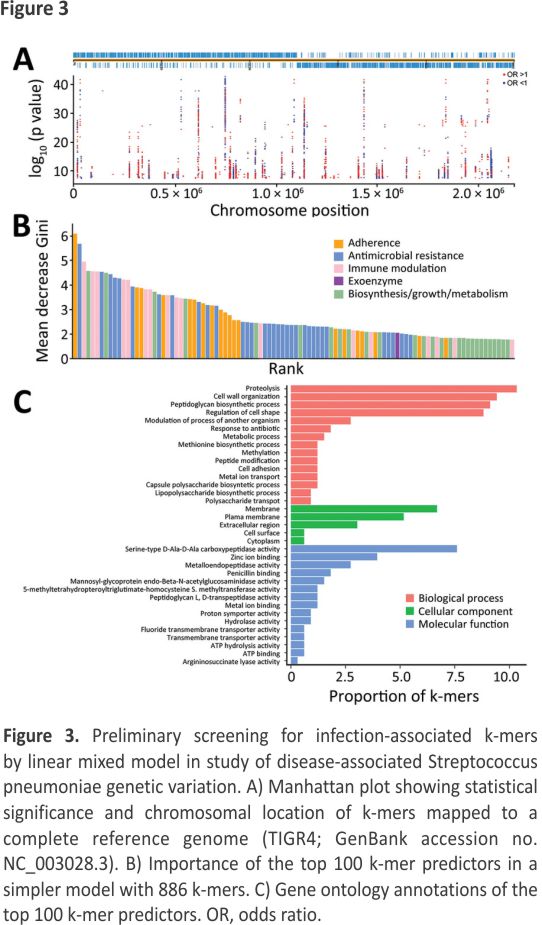
In addition, we sorted the 886 disease-associated k-mers according to estimated importance (Figure 3, panel B). The k-mers were mainly associated with antimicrobial resistance (34%), adherence (20%), immune modulation (17%), and exoenzyme (1%). Moreover, the k-mers were divided into 3 functional gene ontology categories. Among those categories, proteolysis and cell wall organization were the largest subcategories in the biological process, the membrane was the most enriched term in the cellular component, and serine-type D-Ala-D-Ala carboxypeptidase activity was the top term in the molecular function (Figure 3, panel C).
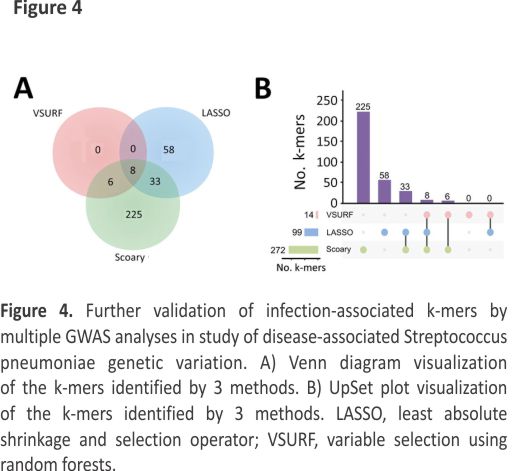
Further Validation of Infection-Associated K-mers by Multiple GWAS Analyses
To reduce the complexity of the model, we used 3 methods to identify consensus infection-associated k-mers (Figure 4). Based on the 886 k-mers screened above, we observed consensus on genomewide statistically significant associations for pathogenicity k-mers; 8 k-mers were identified by all 3 methods. When we used the simplest model with the 8 k-mers, the classification balanced accuracy was 90.89% (95% CI 89.48%– 92.31%) (Table 2). The AUC value was 0.93 (Figure 5, panel A), suggesting that the power of the 8 k-mers to predict disease status was comparable to that of the model with 886 k-mers. Of note, the k-mer predictors still exhibited high classification balanced accuracy in the predominant GPSCs (95.34% for GPSC1 and 92.79% for GPSC321). The importance of the selected k-mers in the final model indicated that these predictors were mainly associated with adherence function (Figure 5, panel B). The highest-ranked predictor (Kmer_9823 in sortase [srtG1]) achieved a classification accuracy of 79.57% on its own and also showed high classification accuracy in the predominant GPSCs (70.04% for GPSC1 and 85.29% for GPSC321). In addition, the best predictor (in srtG1) was associated with GPSC1 and GPSC321 (all p<0.05). For the additional validation analysis that used the best RF classifier k-mer (in srtG1), 2 independent datasets of S. pneumoniae genomes with genotype distribution similar to that of our study were available on the National Center for Biotechnology Information Assembly database (https://www.ncbi.nlm.nih.gov/assemblyExternal Link (data1: 60 noninvasive vs. 60 carriage isolates; data2: 60 invasive versus 60 carriage isolates; the prevalence of the predominant GPSCs [GPSC1 and GPSC321] was 58.3% for noninvasive, 55.0% for invasive and 30.0% for carriage isolates) (Appendix Table 4). Classification accuracy was 75.83% for data1 and 74.17% for data2, similar to that in the larger primary dataset in our study.
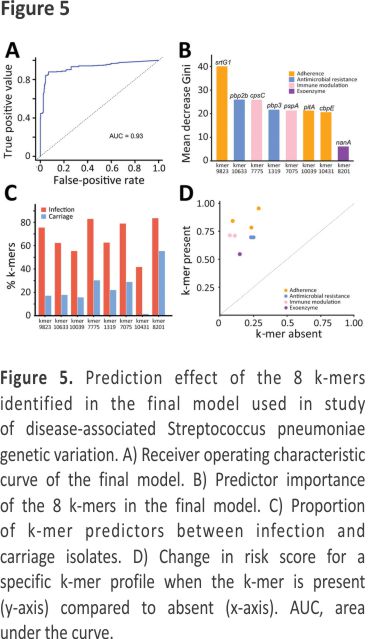
The proportion of k-mers differed significantly between infection and carriage isolates (all p<0.05) (Figure 5, panel C), indicating that the proportion of k-mers was substantially higher in infection isolates than in carriage isolates. The effect of each k-mer on the estimated risk score (Figure 5, panel D), indicated by a point above the diagonal, indicates that the risk score is increased when the k-mer profile is present. The presence of k-mers associated with adherence genes markedly increased the risk for S. pneumoniae infection (odds ratio [OR] 1.88 for Kmer_9823, OR 1.65 for Kmer_10039, and OR 1.69 for Kmer_10431) (Table 3).
Discussion
To explore genomic differences between infection and carriage isolates, linking infection-associated genotypes with disease status is necessary. In our study, the most common serotypes for infection isolates (19F, 6B, 23F) were consistent with the results from other regions of China 18–20 but differed from those from the United States and Japan 21,22. Moreover, we observed considerable ST diversity among infection isolates; the most prevalent genotypes were ST271, ST320, and ST902, a finding consistent with those of previous studies in China but different from those in developed and developing countries 23–25. The resolution of MLST and serotyping for inferring isolate relatedness is limited, so we also used GPSCs to characterize and compare different lineages 26. The most prevalent GPSCs among the infection isolates were GPSC1, GPSC321, and GPSC852, which differed from those in the United States and South Africa 27. Our findings suggest that discrepancies in genotypes on a global scale may be associated with different pathogenicity and evolutionary directions. In our study, associations between specific genotypes (such as 19F and GPSC1) and disease status differed significantly, which is consistent with findings from a study in India 28. Our findings indicate that the presence of specific pathogenic clones may promote infection. In a simple pathogenicity model, all pathogenic clones would belong to specific clusters of genetically related disease-causing isolates (i.e., pathogenic clone hypothesis; Figure 6, panel A), which has been observed for Staphylococcus aureus and S. pneumoniae isolates 29,30. That pathogenicity model is not suitable for all S. pneumoniae clones because many infection isolates clustered in the same branches of the phylogenetic tree as carriage isolates. In addition, traditional genotypes provide little power for identifying small genetic variations at the genomic level 29, suggesting that those genotypes only partially explain the pathogenicity of S. pneumoniae.
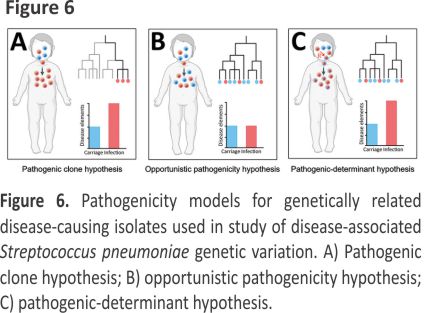
Using high-throughput genome sequencing technologies and bacterial GWAS methods to further explore high-dimensional genetic variation between infection and carriage isolates is essential, thereby revealing the pathogenicity-associated genetic elements of S. pneumoniae. According to the phylogenetic tree, we observed that infection isolates were markedly unevenly distributed across the phylogeny and also clustered with carriage isolates within several lineages, indicating that most lineages are capable of causing infection (i.e., opportunistic pathogenicity hypothesis; Figure 6, panel B). If this hypothesis is reasonable, then GWAS analyses would not detect numerous pathogenicity-associated k-mers. However, the LMM-based GWAS in our study detected 22,790 pathogenicity-associated k-mers. These findings suggest that the enrichment of genetic elements encoding pathogenicity traits may increase the pathogenicity of S. pneumoniae (i.e., pathogenic-determinant hypothesis; Figure 6, panel C), which is consistent with Staphylococcus epidermidis and avian pathogenic Escherichia coli 30,31. In this pathogenic-determinant model, horizontal gene transfer could spread genetic determinants in bacteria such as S. pneumoniae and Klebsiella pneumoniae 32–34, leading various clones to successfully cause disease.
High-throughput genomic data have brought substantial challenges to data analysis because of high-dimensional and highly correlated data structures. In our study, we identified infection-associated k-mers by using a 2-stage comprehensive GWAS analysis process, including LMM for initially screening pathogenic k-mers and multiple GWAS methods for further validation. In the final prediction model, we identified 8 k-mer predictors, which mapped to genes associated with adherence, immune regulation, antibiotic resistance, and exoenzyme. Of the adherence-related genes, srtG1 and the LPxTG-type surface-anchored protein (pitA) are important components of the pneumococcal pilus-2, which plays a crucial role in promoting adhesion, colonization, and cellular invasion 35,36. Classification accuracy of the most important k-mer in srtG1 was high by itself, and that of the additional validation RF analysis based on open datasets was similar, suggesting that this predictor has great potential for predicting pathogenic isolates in a clinical setting. Phosphorylcholine esterase (cbpE) plays an important role in modulating both the phosphorylcholine decoration of its surface and choline-bound surface adhesins, which may contribute to pneumococcal adherence and invasiveness 37. Capsular polysaccharide (CPS) is a major virulence factor in S. pneumoniae. Capsular polysaccharide-protein C (CpsC) has been shown to affect the level of CPS expression and also regulate the assembly, export, and attachment of CPS to the cell wall 38. Pneumococcal surface protein A (PspA) plays a role in preventing complement-mediated opsonization and is also capable of binding to lactoferrin, thereby preventing it from killing pneumococci 39. The infection-associated genes reported in our study (cpsC and pspA) are homologous to the genes associated with invasive pneumococci (cpsA, cpsD, and pspC) identified in previous studies 11,12, providing more evidence for S. pneumoniae pathogenicity. Neuraminidase A encoded by the nanA gene is an essential colonization factor for S. pneumoniae and promotes the growth and survival of the bacteria in the upper respiratory tract 40. Antimicrobial drug use and abuse not only induce widespread multidrug-resistant pneumococci but also increase the susceptibility to invasive disease 41. For decades, penicillin has been the first choice for the treatment of pneumococcal infection, and mutations in penicillin-binding proteins (PBPs) are essential for high-level penicillin resistance 42. Li et al. demonstrated that pbp2b and pbp3 are associated with pneumococcal infection 42. One reason is that PBP2B and PBP3 are involved in the synthesis and growth of bacterial cell walls, which are crucial for the survival and virulence of pneumococci 43. In addition, a previous study revealed a potential association between penicillin resistance and GPSC1 44, and our findings also showed that GPSC1 was associated with pneumococcal infection, suggesting that it cannot support a causal link between resistance and pneumococcal infection and may result from a lineage confounder. In summary, these infection-associated k-mers provide genetic evidence for revealing optimal risk factors for infection isolates, which may offer a theoretical basis for precise targeted interventions.
In this study, we attempted to use the comprehensive analysis strategy to identify pathogenic k-mers by well-characterized S. pneumoniae isolates from a single location so we could reduce the redundancy of k-mer predictors, minimize false-positive associations, and avoid geographic variation. Our consensus findings of pathogenic k-mers from multiple GWAS methods may provide sufficient evidence for clarifying the complex multifactorial pathogenicity of S. pneumoniae. However, among the potential limitations, the first is that S. pneumoniae pathogenesis is a multifactorial and interacting process, but traditional GWAS methods identify the main effect of each genetic variation and ignore the complex gene-gene interactions 45. Therefore, future studies should use the enrichment theory to determine the core functions or pathways for risk genes, which may provide new insights for understanding pathogenesis at functional levels46,47. Second, although k-mers can reflect variation in bacterial genomes, we mapped infection-associated k-mers in our study to reference genomes to identify pathogenic genes, which cannot cover complete genomic variation in the entire species. To overcome those issues, we developed the extended k–mer–based GWAS methods to detect phenotype-specific k-mers without relying on prior annotations or reference genomes 48,49. Third, our study focused mainly on noninvasive rather than invasive isolates, and S. pneumoniae can transition from carriage to infection, suggesting potential similarity in carriage and noninvasive infection isolates. To improve the statistical power and comparability of exploring disease-associated markers, we included infection isolates from children with confirmed associated symptoms and carriage isolates from asymptomatic healthy children.
In conclusion, our 2-stage GWAS analyses identified a subset of 8 pathogenic k-mers associated with adherence, antimicrobial resistance, and immune modulation, indicating that the enrichment of genetic elements encoding pathogenicity traits may increase the pathogenicity of S. pneumoniae. The best predictor for S. pneumoniae infection achieved a high classification accuracy, giving a very simple target for predicting pathogenic isolates in a clinical setting. These findings suggest the complex multifactorial nature of S. pneumoniae pathogenicity and provide genetic evidence for the evolution of virulence and the development of precisely targeted interventions.
Ms. Yang is a graduate student at the Guangdong Pharmaceutical University, in China. Her research interests include pneumococcal disease, evolution of virulence, and genomewide association study.
Acknowledgements
We sincerely thank all study children included in this study. We also thank the research staff and students at Guangdong Pharmaceutical University, China.
This work was supported by the National Natural Science Foundation of China (grant nos. 81973069 and 81602901), the Guangdong Basic and Applied Basic Research Foundation (grant no. 2023A1515011583), and the Key Scientific Research Foundation of Guangdong Educational Committee (grant no. 2022ZDZX2033). The funders had no role in the study design, data collection, and analysis, or interpretation of the data.
References
1. Henriques-Normark B, Tuomanen EI. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harb Perspect Med. 2013;3:a010215.
2. Mancuso G, Midiri A, Gerace E, Biondo C. Bacterial antibiotic resistance: the most critical pathogens. Pathogens. 2021;10: 1310.
3. Subramanian K, Henriques-Normark B, Normark S. Emerging concepts in the pathogenesis of the Streptococcus pneumoniae: From nasopharyngeal colonizer to intracellular pathogen. Cell Microbiol. 2019;21:e13077.
4. Bogaert D, De Groot R, Hermans PW. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect Dis. 2004;4:144–54.
5. Yao KH, Yang YH. Streptococcus pneumoniae diseases in Chinese children: past, present and future. Vaccine. 2008;26:4425 –33.
6. Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008;6:288–301.
7. Piet JR, Geldhoff M, van Schaik BD, Brouwer MC, Valls Seron M, Jakobs ME, et al. Streptococcus pneumoniae arginine synthesis genes promote growth and virulence in pneumococcal meningitis. J Infect Dis. 2014;209: 1781–91.
8. Tunjungputri RN, Mobegi FM, Cremers AJ, van der Gaast-de Jongh CE, Ferwerda G, Meis JF, et al. Phage-derived protein induces increased platelet activation and is associated with mortality in patients with invasive pneumococcal disease. MBio. 2017;8:e01984 –16.
9. Chaguza C, Ebruke C, Senghore M, Lo SW, Tientcheu PE, Gladstone RA, et al. Comparative genomics of disease and carriage serotype 1 pneumococci. Genome Biol Evol. 2022;14:evac052.
10. The CRyPTIC Consortium. Genome-wide association studies of global Mycobacterium tuberculosis resistance to 13 antimicrobials in 10,228 genomes identify new resistance mechanisms. PLoS Biol. 2022;20:e3001755.
11. Lees JA, Ferwerda B, Kremer PHC, Wheeler NE, Serón MV, Croucher NJ, et al. Joint sequencing of human and pathogen genomes reveals the genetics of pneumococcal meningitis. Nat Commun. 2019;10:2176.
12. Obolski U, Gori A, Lourenço J, Thompson C, Thompson R, French N, et al. Identifying genes associated with invasive disease in S. pneumoniae by applying a machine learning approach to whole genome sequence typing data. Sci Rep. 2019;9:4049.
13. Lees JA, Vehkala M, Välimäki N, Harris SR, Chewapreecha C, Croucher NJ, et al. Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes. Nat Commun. 2016;7:12797.
14. Gupta PK. Quantitative genetics: pan-genomes, SVs, and k-mers for GWAS. Trends Genet. 2021; 37: 868–71.
15. Lees JA, Galardini M, Bentley SD, Weiser JN, Corander J. pyseer: a comprehensive tool for microbial pangenome-wide association studies. Bioinformatics. 2018;34: 4310–2.
16. Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: machine learning in Python. J Mach Learn Res. 2011;12:2825–30.
17. Genuer R, Poggi J-M, Tuleau-Malot C. VSURF: an R package for variable selection using random forests. R J. 2015;7:19–33.
18. Geng Q, Zhang T, Ding Y, Tao Y, Lin Y, Wang Y, et al. Molecular characterization and antimicrobial susceptibility of Streptococcus pneumoniae isolated from children hospitalized with respiratory infections in Suzhou, China. PLoS One. 2014; 9:e93 752.
19. Shi W, Li J, Dong F, Qian S, Liu G, Xu B, et al. Serotype distribution, antibiotic resistance pattern, and multilocus sequence types of invasive Streptococcus pneumoniae isolates in two tertiary pediatric hospitals in Beijing prior to PCV13 availability. Expert Rev Vaccines. 2019;18: 89–94.
20. Yu YY, Xie XH, Ren L, Deng Y, Gao Y, Zhang Y, et al. Epidemiological characteristics of nasopharyngeal Streptococcus pneumoniae strains among children with pneumonia in Chongqing, China. Sci Rep. 2019; 9:3324.
21. Suaya JA, Mendes RE, Sings HL, Arguedas A, Reinert RR, Jodar L, et al. Streptococcus pneumoniae serotype distribution and antimicrobial nonsusceptibility trends among adults with pneumonia in the United States, 2009‒2017. J Infect. 2020;81: 557– 66.
22. Yanagihara K, Kosai K, Mikamo H, Mukae H, Takesue Y, Abe M, et al. Serotype distribution and antimicrobial susceptibility of Streptococcus pneumoniae associated with invasive pneumococcal disease among adults in Japan. Int J Infect Dis. 2021;102:260–8.
23. Yan Z, Cui Y, Huang X, Lei S, Zhou W, Tong W, et al. Molecular characterization based on whole-genome sequencing of Streptococcus pneumoniae in children living in southwest China during 2017–2019. Front Cell Infect Microbiol. 2021;11:7267 40.
24. Kellner JD, Ricketson LJ, Demczuk WHB, Martin I, Tyrrell GJ, Vanderkooi OG, et al. Whole-genome analysis of Streptococcus pneumoniae serotype 4 causing an outbreak of invasive pneumococcal disease, Alberta, Canada. Emerg Infect Dis. 2021;27:1867–75.
25. Vorobieva S Jensen V, Furberg AS, Slotved HC, Bazhukova T, Haldorsen B, Caugant DA, et al. Epidemiological and molecular characterization of Streptococcus pneumoniae carriage strains in pre-school children in Arkhangelsk, northern European Russia, prior to the introduction of conjugate pneumococcal vaccines. BMC Infect Dis. 2020;20:279.
26. Gladstone RA, Lo SW, Lees JA, Croucher NJ, van Tonder AJ, Corander J, et al.; Global Pneumococcal Sequencing Consortium. International genomic definition of pneumococcal lineages, to contextualise disease, antibiotic resistance and vaccine impact. EBioMedicine. 2019;43:338–46.
27. Lo SW, Gladstone RA, van Tonder AJ, Lees JA, du Plessis M, Benisty R, et al.; Global Pneumococcal Sequencing Consortium. Pneumococcal lineages associated with serotype replacement and antibiotic resistance in childhood invasive pneumococcal disease in the post-PCV13 era: an international whole-genome sequencing study. Lancet Infect Dis. 2019;19:759 –69.
28. Nagaraj G, Govindan V, Ganaie F, Venkatesha VT, Hawkins PA, Gladstone RA, et al. Streptococcus pneumoniae genomic datasets from an Indian population describing pre-vaccine evolutionary epidemiology using a whole genome sequencing approach. Microb Genom. 2021;7:000645.
29. Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327:469–74.
30. Méric G, Mageiros L, Pensar J, Laabei M, Yahara K, Pascoe B, et al. Disease-associated genotypes of the commensal skin bacterium Staphylococcus epidermidis. Nat Commun. 2018;9:5034.
31. Mageiros L, Méric G, Bayliss SC, Pensar J, Pascoe B, Mourkas E, et al. Genome evolution and the emergence of pathogenicity in avian Escherichia coli. Nat Commun. 2021;12:765.
32. Arnold BJ, Huang IT, Hanage WP. Horizontal gene transfer and adaptive evolution in bacteria. Nat Rev Microbiol. 2022;20: 206–18.
33. Wyres KL, Wick RR, Judd LM, Froumine R, Tokolyi A, Gorrie CL, et al. Distinct evolutionary dynamics of horizontal gene transfer in drug resistant and virulent clones of Klebsiella pneumoniae. PLoS Genet. 2019; 15:e1008114.
34. Salvadori G, Junges R, Morrison DA, Petersen FC. Competence in Streptococcus pneumoniae and close commensal relatives: mechanisms and implications. Front Cell Infect Microbiol. 2019; 9:94.
35. Bagnoli F, Moschioni M, Donati C, Dimitrovska V, Ferlenghi I, Facciotti C, et al. A second pilus type in Streptococcus pneumoniae is prevalent in emerging serotypes and mediates adhesion to host cells. J Bacteriol. 2008;190: 5480 –92.
36. Dzaraly ND, Muthanna A, Mohd Desa MN, Taib NM, Masri SN, Rahman NIA, et al. Pilus islets and the clonal spread of piliated Streptococcus pneumoniae: A review. Int J Med Microbiol. 2020; 310:151449.
37. Hermoso JA, Lagartera L, González A, Stelter M, García P, Martínez-Ripoll M, et al. Insights into pneumococcal pathogenesis from the crystal structure of the modular teichoic acid phosphorylcholine esterase Pce. Nat Struct Mol Biol. 2005;12: 533–8.
38. Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008; 6:288–301.
39. Marquart ME. Pathogenicity and virulence of Streptococcus pneumoniae: Cutting to the chase on proteases. Virulence. 2021; 12:766–87.
40. Brittan JL, Buckeridge TJ, Finn A, Kadioglu A, Jenkinson HF. Pneumococcal neuraminidase A: an essential upper airway colonization factor for Streptococcus pneumoniae. Mol Oral Microbiol. 2012;27:270–83. 41. Navarro-Torné A, Dias JG, Hruba F, Lopalco PL, Pastore-Celentano L, Gauci AJ; Invasive Pneumococcal Disease Study Group. Risk factors for death from invasive pneumococcal disease, Europe, 2010. Emerg Infect Dis. 2015;21: 417–25.
42. Li Y, Metcalf BJ, Chochua S, Li Z, Gertz RE Jr, Walker H, et al. Penicillin-binding protein transpeptidase signatures for tracking and predicting β-lactam resistance levels in Streptococcus pneumoniae. MBio. 2016;7: e00756–16.
43. Gibson PS, Veening JW. Gaps in the wall: understanding cell wall biology to tackle amoxicillin resistance in Streptococcus pneumoniae. Curr Opin Microbiol. 2023;72:102261.
44. Egorova E, Kumar N, Gladstone RA, Urban Y, Voropaeva E, Chaplin AV, et al. Key features of pneumococcal isolates recovered in Central and Northwestern Russia in 2011-2018 were determined through whole-genome sequencing. Microb Genom. 2022;8:mgen 000851.
45. Bai G, Vidal JE. Editorial: molecular pathogenesis of Pneumococcus. Front Cell Infect Microbiol. 2017; 7:310.
46. Chen L, Zhang YH, Wang S, Zhang Y, Huang T, Cai YD. Prediction and analysis of essential genes using the enrichments of gene ontology and KEGG pathways. PLoS One. 2017;12:e0184129.
47. Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587–92.
48. Jaillard M, Lima L, Tournoud M, Mahé P, van Belkum A, Lacroix V, et al. A fast and agnostic method for bacterial genome-wide association studies: Bridging the gap between k-mers and genetic events. PLoS Genet. 2018;14:e 1007758.
49. Aun E, Brauer A, Kisand V, Tenson T, Remm M. A k-mer-based method for the identification of phenotype-associated genomic biomarkers and predicting phenotypes of sequenced bacteria. PLOS Comput Biol. 2018;14:e1006434.
CREDITS: Yang S, Chen J, Fu J, et al. Disease-Associated Streptococcus pneumoniae Genetic Variation. Emerging Infectious Diseases. 2024; 30(1):39-49. doi:10.3201/eid3001. 221927.
: an evolving threat to the Nigerian health system – a review")



: an evolving threat to the Nigerian health system – a review")

