

Athina Spiliopoulou, Andrii Iakovliev, Darren Plant, Megan Sutcliffe, Seema Sharma, Cankut Cubuk, Myles Lewis, Costantino Pitzalis, Anne Barton, Paul M. McKeigue
Abstract
Objective
The sparse effector “omnigenic” hypothesis postulates that the polygenic effects of common single nucleotide polymorphisms (SNPs) on a typical complex trait are mediated by trans effects that coalesce on expression of a relatively sparse set of core genes. The objective of this study was to identify core genes for rheumatoid arthritis by testing for association of rheumatoid arthritis with genome-wide aggregated trans effects (GATE) scores for expression of each gene as transcript in whole blood or as circulating protein levels.
Methods
GATE scores were calculated for 5,400 cases and 453,705 non-cases of primary rheumatoid arthritis in UK Biobank participants of European ancestry.
Results
Testing for association with GATE scores identified 16 putative core genes for rheumatoid arthritis outside the HLA region, of which six—TP53BP1, PDCD1, TNFRSF14, LAIR1, LILRA4, and IDO1—were supported by Mendelian randomization analysis based on the marginal likelihood of the causal effect parameter. Five of these 16 genes were validated by a reported association of rheumatoid arthritis with SNPs within 200 kb of the transcription site, eight by association of the measured protein level with rheumatoid arthritis in UK Biobank, 10 by experimental perturbation in mouse models of inflammatory arthritis, and two—CTLA4 and PDCD1—by evidence that drugs targeting the gene cause or ameliorate inflammatory arthritis in humans. Fourteen of these 16 genes are in pathways affecting immunity or inflammation, and six— CD5, CTLA4, TIGIT, LAIR1, TNFRSF14, and PDCD1—encode receptors that have been characterized as immune checkpoints exploited by cancer cells to escape the immune response.
Conclusion
These results highlight the key role of immune checkpoints in rheumatoid arthritis and identify possible therapeutic targets.
INTRODUCTION
Rheumatoid arthritis is an immune-mediated inflammatory disease with a prevalence of about 1% in most populations. The genetic information for discrimination, equal to the logarithm to base two of the recurrence risk ratio in first-degree relatives,1 is about 2.3 bits,2 of which the HLA region accounts for about 0.8 bits.3 Although the catalog of genome-wide association studies (GWAS) lists 337 genomic regions outside the HLA region that contain single nucleotide polymorphisms (SNPs) associated with rheumatoid arthritis at the conventional genome-wide threshold of P < 5 × 10−8, the genes in these regions are mostly broadly expressed and are not in pathways specifically relevant to immune-mediated disease.4 Although the treatment of rheumatoid arthritis has been revolutionized by drugs that target specific proteins mediating inflammation, the contribution of GWAS to discovery of drug targets for rheumatoid arthritis has been limited.5
Conventional GWAS analyses focus on identifying the nearby genes (usually within 200 kb) that mediate the effects of common variants on disease through cis effects. However, for a typical gene, most of the SNP heritability of levels of the transcript or encoded protein is attributable to trans effects of variants that are distant from the transcription site.6 On this basis, the “omnigenic” sparse effector model was proposed as a fundamental rethink of the genetic architecture of complex traits.7 This postulates that most of the polygenic effects on a typical complex trait are mediated through weak trans effects of common variants that coalesce on expression of a relatively sparse set of “core” effector genes in relevant tissues. The availability of summary statistics from large GWAS studies of transcripts in whole blood or proteins in plasma has made it possible to test this hypothesis by constructing genotypic predictors of gene expression based on aggregated trans effects and testing these genotypic scores for association with the disease or trait under study. We have reported an application of this genome-wide aggregated trans effects (GATE) analysis pipeline to type 1 diabetes, which identified a set of putative core genes regulating the differentiation and activity of CD4+ Treg cells.8 The objective of this study was to investigate whether GATE analysis can identify core genes for rheumatoid arthritis.
MATERIALS AND METHODS
Ethical approval
Ethical approval for the UK Biobank was previously obtained from the North West Centre for Research Ethics Committee (11/ NW/0382). Informed consent was obtained for all study participants. The work described herein was approved by the UK Biobank under application 23652. Ethical approval for the transcriptomics study was granted by the North West 6 Central Manchester South Research Ethics Committee (COREC 04/ Q1403/37). All patients provided written consent.
Case definition
The case definition of rheumatoid arthritis in the UK Biobank cohort was based on any of three criteria: (1) hospital discharge or death certificate with an International Classification of Diseases diagnostic code for rheumatoid arthritis; (2) any primary care diagnosis of rheumatoid arthritis; or (3) self-reported diagnosis of rheumatoid arthritis supported by prescription of a disease-modifying antirheumatic drug at baseline or during follow-up. Of 487,152 individuals with nonmissing phenotype and genotype data, 5,958 had ever been diagnosed with rheumatoid arthritis. When those with a diagnosis of Sjögren’s disease antedating the diagnosis of rheumatoid arthritis were excluded, 5,731 cases of primary rheumatoid arthritis remained. The full dataset was pruned to ensure that no pairs of individuals with kinship coefficient >0.05 remained. As summary statistics for cis and trans effects of SNP genotypes on gene expression were available only from studies of individuals of European ancestry, the dataset for trans effects analysis was restricted to those participants who reported their ethnic origin as white. After applying these exclusions, there were 451,447 individuals, of whom 5,292 were classified as cases. Of these, 2,015 were classified as incident cases, on the basis that the first mention of a diagnosis of rheumatoid arthritis was later than the date on which they were assessed.
GATE analysis
Methods for GATE analysis have been described previously.8 The data sources and analytical steps are illustrated in Figure 1. The list of SNPs that were typed or imputed in UK Biobank was uploaded to the GENOSCORES server. A database query extracts the univariate coefficients of regression of expression of each target gene on each of these SNPs, filtered by setting a threshold of P < 10−5. For each target gene and each clump of associated SNPs containing at least one SNP associated with expression of the gene at P < 10−6, the vector of coefficients is multiplied by the inverse correlation matrix among SNP genotypes computed from the 1000 Genomes reference panel to obtain a vector of multi- variable weights that are corrected for linkage disequilibrium (LD). A pseudoinverse solution is implemented to handle ill-conditioned matrices in clumps with highly correlated SNPs. For each gene and each clump of associated SNPs, a locus-specific score is computed by multiplying the vector of adjusted weights by the matrix of SNP genotypes in the target dataset. The locus-specific scores for each gene are summed over trans-quantitative trait loci (QTLs) to obtain genome-wide aggregated trans scores.
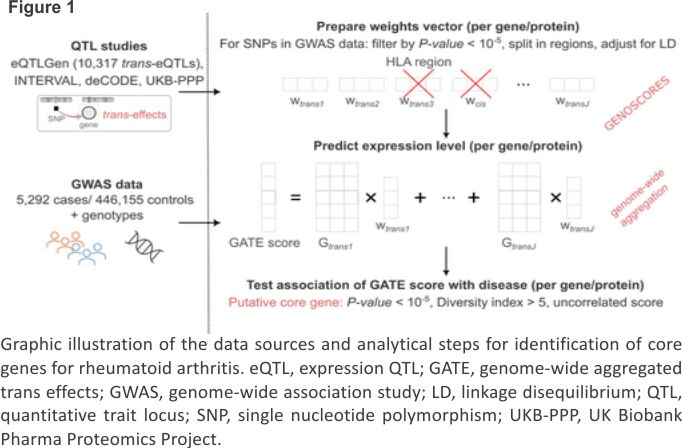
Trans-expression QTL (trans-eQTL) scores were computed from eQTLGen phase 1, in which only 10,317 trait-associated SNPs were tested for trans associations.9 For these scores, the threshold for defining clumps of associated SNPs was relaxed to P < 10−5 because for these trait-associated SNPs, the prior probability of an effect on gene expression is higher than it is for random SNPs. Trans-protein QTL (trans-pQTL) scores were computed from three studies of circulating proteins:
- 1,478 proteins on the SomaLogic panel measured in plasma on 3,301 blood donors in the INTERVAL study.10
- 4,719 proteins on the SomaLogic version 4 panel measured in plasma on 35,559 Icelanders in the deCODE study11; 2,207 aptamers on this platform that appeared to cross-react with CFH, encoding complement factor H, were excluded. The criteria for identifying these aptamers were a trans-pQTL at the CFH locus, no cis-pQTL, and association of the trans score with age-related macular degeneration.
2,923 proteins on the Olink Explore panel measured in plasma on 54,306 participants in UK Biobank.12
As the HLA region is a hotspot for trans-QTLs for genes involved in immunity and inflammation13 and associations of these trans-QTLs with autoimmune disease are heavily confounded by the direct effects of HLA antigens, the HLA region (from 25 to 34 Mb on chromosome 6) was excluded from the computation of genome-wide trans scores. Cis-eQTLs and cis-pQTLs were excluded from the aggregated trans scores and tested for association with the disease separately.
Statistical analysis
When testing for association with genotypic scores for Olink proteins calculated from the UK Biobank proteomics study, the 54,306 participants who were included in the proteomics study were excluded from tests of association of the genotypic scores with the outcome. A logistic regression model was fitted with rheumatoid arthritis as response variable with sex and the first 20 genotypic principal components as covariates. The fitted values from this null model were used to compute tests for association with the cis score and aggregated trans score predicting the levels of a transcript or circulating protein. These tests were computed as efficient score tests based on the gradient and second derivative of the log-likelihood at the null. Log odds ratios in the tables have been standardized by multiplying them by the SD of the score so that the coefficient is the log odds ratio associated with an increase of the score by one SD.
As an index of the effective number of unlinked trans-QTLs contributing to each genome-wide trans score, we calculated the diversity index or Hill number.14 For each gene, the diversity index was computed from the variances σ1,…,σK of the K locus–specific trans scores as 2−∑ipi log2pi, where pi = σi2 / ∑j σj2. This index can take values from 1, if one of the QTLs has much larger variance than the others, to K, if the variances of the locus-specific trans scores are equal.
For each putative core gene identified through aggregating the effects of at least 10 trans-QTLs, an instrumental variable (“Mendelian randomization”) analysis was undertaken. This was based on constructing a scalar genetic instrument from each trans-QTL and marginalizing over the distribution of direct (pleiotropic) effects of the instrument on the outcome to compute the likelihood of the causal effect parameter as described elsewhere.15 Cis-QTLs were excluded from these analyses because cis-acting SNPs frequently alter the splicing of the gene product so that effects on the measured level of transcript or circulating protein do not correspond to effects on function. A null result in these Mendelian randomization analyses does not exclude a causal effect because even with 10 or more instruments (trans-QTLs), there may not be enough information in the data to learn the posterior distribution of direct effects.
Filtering and validation criteria
As before,8 the GATE scores were filtered to retain only those scores for which the effective number of trans-QTLs was greater than five. This retained 413 aggregated trans-eQTL scores for 413 unique genes and 3,256 aggregated trans-pQTL scores for 2,586 unique genes. For the initial list of putative core genes, we set a threshold of P < 10−5 for association of disease with GATE scores.
We defined six criteria for validation as a core gene:
1. Any SNP association with rheumatoid arthritis, reported in the GWAS catalog, at the conventional threshold of P < 5 × 10−8 within 200 kb of the transcription site of the target gene. As cis-SNPs were excluded from the GATE score, this is orthogonal validation. The 200-kb cutoff is based on the distribution of distances from protein QTL variants to transcription start site.16 Although this criterion would not be appropriate for identifying the gene that mediates the cis effect of a disease-associated SNP, if the gene has already been identified through trans effects as a putative core gene for the disease and there is a SNP association near its transcription site, then this gene is the most likely mediator of the cis effect.
2. Instrumental variable analysis (“Mendelian randomization”), based on marginalizing over the distribution of pleiotropic effects of trans-QTLs, supports a causal effect of the transcript or protein at P < 0.01. This criterion is evaluated only if the target gene has at least 10 trans-QTLs because, otherwise, there is not enough information to infer the distribution of pleiotropic effects.
3. Association of disease with the measured level of the encoded protein in the UK Biobank proteomics study, in the same direction as the association with the aggregated trans score, with magnitude (standardized log odds ratio) of the protein association at least twice that of the trans score association. Evidence of causality is strengthened if levels of the protein are associated with incident rheumatoid arthritis, excluding those already diagnosed at baseline.
4. Drugs targeting the gene product, its ligand, or its receptor cause inflammatory arthritis or have shown efficacy against rheumatoid arthritis in a phase 2 trial.
5. Perturbation of the gene by knockout, transduction, or over-expression, or perturbation of the gene product by an inhibitor or an agonist, alters the severity of disease in an experimental model of inflammatory arthritis.
6. Rare variants in the gene cause a monogenic form of the disease. From searching PubMed, 11 genes were identified as reported monogenic causes of inflammatory arthritis: LACC1, LRBA, NFIL3, UNC13D, NOD2, NLRP3, MEFV, TNFAIP3, PRF1, STX11, ACP5.17-21
RESULTS
Trans-eQTL scores
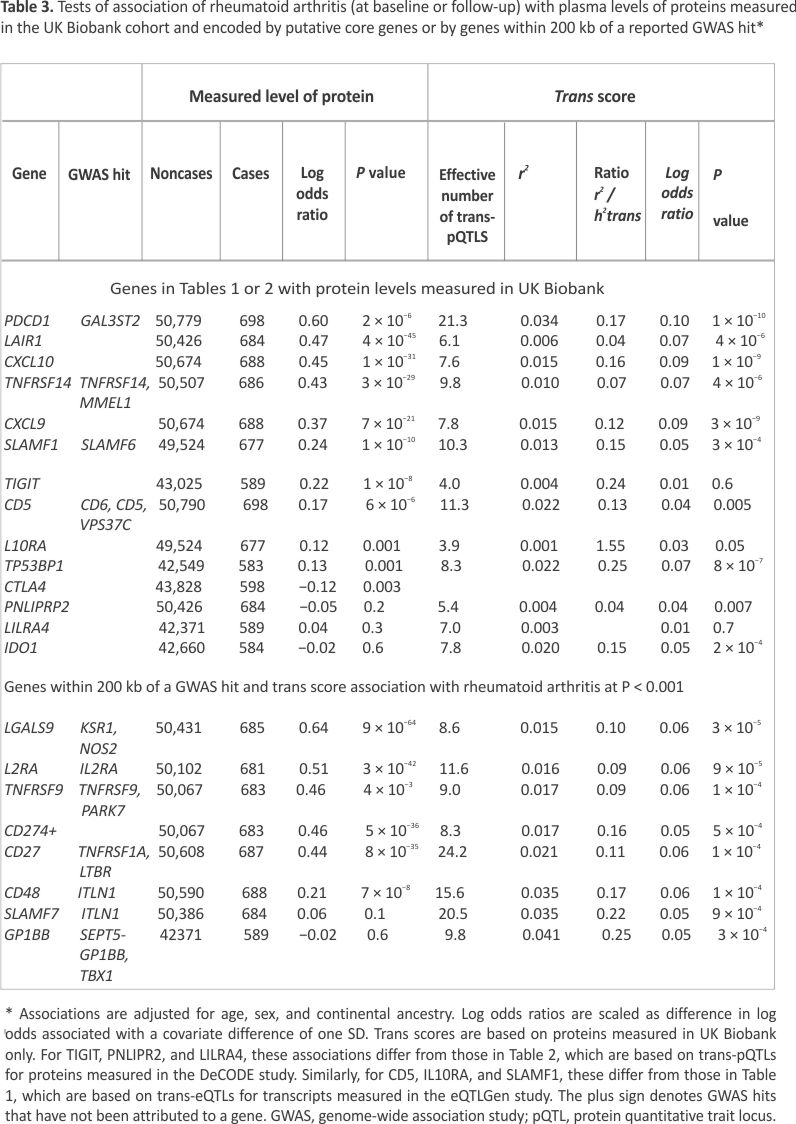
Table 1 shows the six putative core genes identified through association with aggregated trans-eQTL scores. For three of these genes—CD5, CTLA4, and SLAMF1—associations of rheumatoid arthritis with SNPs within 200 kb of the transcription site are listed in the GWAS catalog. For these SNP associations, gene names recorded in the GWAS catalog in the fields “Reported Gene” or “Mapped Gene” are shown in the last column of Table 1. As previously reported for type 1 diabetes,8 the cis-eQTL association of CTLA4 is in the opposite direction to the association with the aggregated trans score; this may be explained by cis-acting SNPs that alter the splicing of this gene.
 Figure 2 shows that the aggregated trans-eQTL scores for the six putative core genes are only weakly correlated, indicating that they do not share most of their trans-eQTLs. Supplementary Table S1 shows that multiple regions containing SNPs previously identified as associated with rheumatoid arthritis are trans-eQTLs for one or more of these putative core genes. Notably, seven regions outside the HLA region that contain SNPs associated with rheumatoid arthritis are trans-eQTLs for CTLA4.
Figure 2 shows that the aggregated trans-eQTL scores for the six putative core genes are only weakly correlated, indicating that they do not share most of their trans-eQTLs. Supplementary Table S1 shows that multiple regions containing SNPs previously identified as associated with rheumatoid arthritis are trans-eQTLs for one or more of these putative core genes. Notably, seven regions outside the HLA region that contain SNPs associated with rheumatoid arthritis are trans-eQTLs for CTLA4.
![]() To test whether genetic effects on the proportions of immune cell types could explain the associations of rheumatoid arthritis with trans scores for predicted gene expression, we used summary statistics from the SardiNIA study of immune cell phenotypes in peripheral blood22 to calculate genome-wide scores for each immune cell phenotype. We tested these scores for association with rheumatoid arthritis. Supplementary Table S2 shows that rheumatoid arthritis was associated inversely with the proportion of CD4+ T cells that were Treg cells and positively associated with the absolute count of CD4+ T cells. Although CTLA4 is expressed on CD4+ T cells, the association of rheumatoid arthritis with the polygenic score for CD4+ T cell count was much weaker than the association with the GATE score for CTLA4.
To test whether genetic effects on the proportions of immune cell types could explain the associations of rheumatoid arthritis with trans scores for predicted gene expression, we used summary statistics from the SardiNIA study of immune cell phenotypes in peripheral blood22 to calculate genome-wide scores for each immune cell phenotype. We tested these scores for association with rheumatoid arthritis. Supplementary Table S2 shows that rheumatoid arthritis was associated inversely with the proportion of CD4+ T cells that were Treg cells and positively associated with the absolute count of CD4+ T cells. Although CTLA4 is expressed on CD4+ T cells, the association of rheumatoid arthritis with the polygenic score for CD4+ T cell count was much weaker than the association with the GATE score for CTLA4.
Trans-pQTL scores
Table 2 shows the 10 putative core genes identified through association with aggregated trans-pQTL scores. The strongest GATE score association with rheumatoid arthritis is for PDCD1, which encodes the immune checkpoint receptor programmed cell death protein 1 (PD-1). Supplementary Table S3 shows that the trans-pQTLs contributing to this GATE score include 12 regions containing genes previously associated with rheumatoid arthritis through GWAS studies: SDF4, AP4B1, PTPN22, CTLA4, BACH2, TRAF1, IL2RA, TSPAN14, PTPN11, ILF3, FUT2 and AIRE. This coalescence of trans effects of disease-associated SNPs on expression of a disease-relevant target gene is consistent with the sparse effector hypothesis.
![]() Supplementary Figure S1 shows that the aggregated trans-pQTL scores for seven of these putative core genes are correlated, indicating that they share trans-pQTLs. The highest correlation (0.6) is between the scores for CXCL10 and CXCL9. Supplementary Table S3 shows that these two genes share nine trans-pQTL regions containing genes —PTPN22, GCKR, IL18R1, STAT4, CCR1, IFNG-AS1, PTPN11, PTPN2, and TYK2—previously identified as associated with rheumatoid arthritis. This sharing of trans-QTLs means that we cannot distinguish, without other evidence, which of these two target genes is most likely to be causal.
Supplementary Figure S1 shows that the aggregated trans-pQTL scores for seven of these putative core genes are correlated, indicating that they share trans-pQTLs. The highest correlation (0.6) is between the scores for CXCL10 and CXCL9. Supplementary Table S3 shows that these two genes share nine trans-pQTL regions containing genes —PTPN22, GCKR, IL18R1, STAT4, CCR1, IFNG-AS1, PTPN11, PTPN2, and TYK2—previously identified as associated with rheumatoid arthritis. This sharing of trans-QTLs means that we cannot distinguish, without other evidence, which of these two target genes is most likely to be causal.
To investigate whether these results were replicated in independent datasets, we searched the “expression quantitative trait score” (eQTS) summary statistics released by the eQTLGen Consortium.9 Their study used summary statistics from two published GWAS of rheumatoid arthritis to construct polygenic risk scores that were tested for association with gene expression in an individual-level dataset. Supplementary Table S4 shows that for three of the genes in Tables 1 or 2—PDCD1, TIGIT, and CTLA4—expression levels were associated with polygenic scores for rheumatoid arthritis.
Nearby GWAS hits
For five of the 16 putative core genes—CD5, CTLA4, SLAMF1, PDCD1, and TNFRSF14—that were identified through association of GATE scores with rheumatoid arthritis, associations of rheumatoid arthritis with SNPs within 200 kb of the transcription site are listed in the GWAS catalog. The attribution of the GWAS hit to the target gene is supported by a cis-QTL association for CTLA4. For CD5, CTLA4, and TNFRSF14, the target gene is included in the GWAS catalog as one of the attributed genes. The association with SNPs in the SLAMF1 region had been attributed to SLAMF6, and the association with SNPs in the PDCD1 region had been attributed to GAL3ST2, 40 kb upstream of PDCD1.
Mendelian randomization
Supplementary Table S5 shows that of the 10 putative core genes identified through aggregated trans effects on protein levels, six were supported at P < 0.01 by a Mendelian randomization analysis based on computing the marginal likelihood of the causal effect parameter.15 Scatter plots of the coefficients for the effects of the trans-pQTLs are shown for LAIR1, TP53BP1, TNFRSF14, and PDCD1 in Supplementary Figures S2–S5. The coefficients for the effects of the pQTLs on rheumatoid arthritis are plotted against the coefficients for the effects of the pQTLs on circulating levels of the protein. The slope of the straight line shown in each plot is the maximum likelihood estimate of the causal effect parameter based on marginalizing over the posterior distribution of the direct (pleiotropic) effects. If there were no direct effects of the trans-pQTLs on rheumatoid arthritis and no uncertainty in the estimates of coefficients of regression of disease and protein levels on the trans-pQTLs, the points in this plot would lie on a straight line through the origin, and the slope of this line would be the causal effect parameter. We emphasize that Mendelian randomization analysis has limitations and that the results should be interpreted as only one line of evidence to be combined with evidences from other sources.
Associations with measured levels of proteins and transcripts
Of the 54,306 individuals in the UK Biobank proteomics study, 688 had a diagnosis of primary rheumatoid arthritis at baseline or during follow-up. Table 3 shows that of the 14 putative core genes for which the protein encoded by the gene was measured in the UK Biobank proteomics study, nine met the criterion of association in the same direction as the trans score effect with a standardized log odds ratio at least twice as high for the measured protein as for the GATE score that predicts the protein level. Because these associations of protein levels with rheumatoid arthritis include cases already diagnosed, they may be altered by the effects of the disease or by drug treatment.
 The lower part of Table 3 shows eight additional candidates for core gene status identified as genes within 200 kb of a GWAS hit that have a GATE score associated with rheumatoid arthritis at the less stringent threshold of P < 0.001 (on the basis that a nearby GWAS hit increases the prior probability of a causal effect). For five of these eight genes—LGALS9, IL2RA, TNFRSF9, CD274, and CD27—levels of the encoded protein were strongly associated with rheumatoid arthritis. Supplementary Table S6 shows that for 10 of the 14 proteins associated with rheumatoid arthritis at baseline or follow-up at a standardized log odds ratio >0.2, these associations persisted after restriction to incident cases diagnosed during follow-up.
The lower part of Table 3 shows eight additional candidates for core gene status identified as genes within 200 kb of a GWAS hit that have a GATE score associated with rheumatoid arthritis at the less stringent threshold of P < 0.001 (on the basis that a nearby GWAS hit increases the prior probability of a causal effect). For five of these eight genes—LGALS9, IL2RA, TNFRSF9, CD274, and CD27—levels of the encoded protein were strongly associated with rheumatoid arthritis. Supplementary Table S6 shows that for 10 of the 14 proteins associated with rheumatoid arthritis at baseline or follow-up at a standardized log odds ratio >0.2, these associations persisted after restriction to incident cases diagnosed during follow-up.
Table 3 shows that the squared correlations (r2) between the GATE scores and the measured protein levels are typically only about 2% and that the proportion of variance explained by the GATE score is typically 10% to 20% of the total SNP heritability attributable to trans effects (h2trans) estimated previously in the UK Biobank proteomics dataset.12 If the association of the protein with disease is causal, the effect size associated with the measured protein level should be about seven times the effect size associated with the GATE score (the ratio of effect sizes scales with the correlation of the score with the measured value). For the top five genes in Table 3, the ratio of measured protein effect size to GATE score effect size is between four and seven, broadly consistent with this prediction.
To compare whole blood transcript levels in cases of rheumatoid arthritis and healthy controls, we examined two microarray studies: a published study of 66 cases of rheumatoid arthritis not yet treated with a conventional synthetic disease-modifying antirheumatic drug (csDMARD) and 35 healthy controls in Keio, Japan23 and an unpublished study of 55 csDMARD-treated cases of rheumatoid arthritis and 10 healthy controls in Manchester, England. Supplementary Table S7 shows that expression levels of CD5 and LAIR1 were inversely associated with rheumatoid arthritis in both studies. These associations with transcript levels were opposite in direction to the associations with the soluble proteins in UK Biobank, consistent with the possibility that the soluble proteins act as decoys to down-regulate the cellular receptor. We emphasize that to characterize case-control differences in whole blood transcript levels will require larger studies using methods that distinguish splice variants and control for cell-type proportions.
Summary of validation
Supplementary Table S8 tabulates the genes in Tables 1, 2, or 3 for which experimental perturbation has demonstrated an effect in a mouse model of inflammatory arthritis. For two of these genes, there is at least preliminary clinical evidence of efficacy of drugs targeting the encoded protein against rheumatoid arthritis. Table 4 summarizes the results of applying five criteria for validation of each putative core gene, excluding the sixth criterion of monogenic disease, which was not met by any of these genes. The strongest evidence is for PDCD1, which meets all five criteria: nearby GWAS hit, protein association, Mendelian randomization support, validation from perturbation in an experimental model, and effects of a targeted agonist against rheumatoid arthritis in a phase 2 trial.
 DISCUSSION
DISCUSSION
From aggregated trans effects, this study identified 16 genes outside the HLA region as putative core genes for rheumatoid arthritis. Six of these—CD5, CTLA4, TIGIT, LAIR1, PDCD1, and TNFRSF14 —encode proteins that have been identified as immune checkpoints, defined broadly as receptors on immune cells that are exploited by cancer cells to escape the immune response.24-27 Inflammatory arthritis and other autoimmune reactions are common adverse effects of inhibitors of these immune checkpoints used in cancer therapy. LILRA4 encodes leukocyte immunoglobin-like receptor subfamily A member 4, a cell surface protein expressed on plasmacytoid dendritic cells; it has been proposed that LILRA4 is an “innate checkpoint,” analogous to the checkpoints identified in the adaptive immune system.28
For 10 of these genes, experimental perturbation in a mouse model has been shown to influence inflammatory arthritis.29-38 For two putative core genes— CTLA4 and PDCD1—there is at least preliminary evidence of efficacy against rheumatoid arthritis in clinical trials of drugs that target the protein.39, 40
Of the six immune checkpoint genes identified through GATE analysis as putative core genes for rheumatoid arthritis, GATE scores for four—CD5, CTLA4, PDCD1, and TIGIT—are associated with type 1 diabetes also8 (result for PDCD1 to be reported elsewhere). The other two—LAIR1 and TNFRSF14 —appear to be more specifically associated with rheumatoid arthritis. TNFRSF14 was originally identified as a receptor for glycoprotein D of herpes simplex virus; it is now considered to be an immune checkpoint that inhibits immune response by binding to B- and T-lymphocyte attenuator, encoded by BTLA, and to CD160, an Ig-like glycoprotein expressed on γδ T cells. A TNFRSF14-Ig fusion protein aggravated autoimmune arthritis in the collagen-induced arthritis mouse model.34
Of the 16 putative core genes identified through GATE analysis, 11 are expressed specifically by immune cells, mostly T cells. The genes not expressed specifically by immune cells are IDO1, FBLN7, STAP1, TP53BP1, and PNLIPRP2. IDO1 modulates immune checkpoint– mediated immunosuppression by mechanisms that are not yet clear.41 STAP1 has been reported to up-regulate activation of T cells via the T cell receptor.42 For FBLN7 and TP53BP1, there is some experimental support for a role in inflammatory arthritis. FBLN7 encodes the extracellular matrix protein fibulin-7, which binds to human monocytes via integrins α5β1 and α2β1; in a mouse model of systemic inflammation, it inhibits chemokine-mediated migration of macrophages.43 TP53BP1 encodes the p53-binding protein 1 that is required for accumulation of p53 to facilitate repair of DNA double-strand breaks. Knockout of the gene encoding p53 in mice increases disease severity in the collagen-induced arthritis model.32 PNLIPRP2, which is only just above the P-value threshold of 10−5 and has no support from association of measured protein levels with disease, may be a chance finding.
Six additional candidates for core gene status are identified by the combination of a GATE score association with rheumatoid arthritis at P < 0.001, a reported GWAS hit within 200 kb of the transcription site, and association of measured protein levels with rheumatoid arthritis. For five of these genes — LGALS9, IL2RA, TNFRSF9, CD274, and CD27—there is experimental validation from perturbation in a mouse model of inflammatory arthritis.38,44-47 For three of these five genes, the GWAS hit had been attributed to another nearby gene or not attributed at all. The association with CD274 that encodes the ligand for the PD-1 receptor is further support for a key role of PD-1 signaling in rheumatoid arthritis. LGALS9 encodes galectin-9, one of four ligands for the immune check- point receptor TIM-3, also known as hepatitis A virus cellular receptor 2; blockade of TIM-3 ameliorates arthritis in a mouse model.48
The trans-pQTL scores calculated in this study are for the predicted level of the circulating protein. In cases where this protein is the soluble form of a cellular receptor, the associations of disease with levels of the soluble form are not necessarily in the same direction as the associations with expression of the receptor. Thus, for PD-1, the measured level of the soluble protein and the GATE score for the protein level are positively associated with rheumatoid arthritis. This effect is in the opposite direction to what would be predicted from clinical experience with antagonists and agonists of the PD-1 receptor. A possible explanation for this is that the soluble protein acts as a decoy for the ligand for the cellular receptor.49, 50
Strengths of this study are that the cases and non-cases are from a single cohort, that diagnoses based on primary care records or self-report are validated by drug prescriptions, and that the design effectively rules out reverse causation because the background prevalence of rheumatoid arthritis in the population samples from which genetic scores were constructed was only about 1%. The main limitation of this study is that for genetic prediction from trans effects on gene expression in whole blood, it relies on eQTLGen phase 1, in which only 10,316 trait-associated SNPs were tested for trans associations. Another limitation is that the eQTLGen summary statistics were not directly adjusted for cell-type proportions in each blood sample, although they were adjusted for principal components as a proxy for cell-type proportions. We addressed this by examining the associations of rheumatoid arthritis with polygenic scores for immune cell phenotypes; CD4+ T cells were the only cell type for which a polygenic score was positively associated with rheumatoid arthritis. As the GATE score associations with rheumatoid arthritis do not match the expression profile of this cell type, it is unlikely that confounding by cell-type proportions can explain these associations. These limitations will be overcome when more comprehensive summary statistics are available from the whole blood transcriptomics study that is now planned for a subset of the UK Biobank cohort. For trans-pQTLs in whole blood, comprehensive summary statistics on SNP associations are available for 2,923 proteins on the Olink platform measured on 54,000 participants in UK Biobank, but even with this large sample size, the GATE scores typically explain only about 10% of the estimated SNP heritability attributable to trans effects. We are nevertheless able to detect associations of these GATE scores with disease because the underlying associations, estimated from the associations of measured protein levels with disease, are typically strong.
Where variants with pleiotropic effects on gene expression have large effects on disease risk, GATE analysis alone cannot reliably distinguish causal genes from genes that are regulated by these pleiotropic variants. For this reason, we excluded trans-QTLs in the HLA region from the computation of GATE scores, as in our earlier study of type 1 diabetes. Where variants have large direct effects on disease risk, the eQTS method9 is expected to be less powerful than GATE analysis because such variants dilute the association of the target gene with the polygenic risk score. This may explain why only three of the 16 putative core genes were replicated in the eQTS summary statistics, even though the eQTS analysis was not limited to the 10,316 SNPs used in eQTLGen phase 1.
The coalescence of disease-associated SNP effects on expression of a small number of target genes, demonstrated in this study, provides broad support for the sparse effector hypothesis, at least for immune-mediated inflammatory disease. As disease-relevant genes are enriched with redundant enhancer domains and depleted of cis-eQTLs of large effect,51,52 they are often not detected by a conventional SNP-by-SNP GWAS analysis. Whatever theoretical questions remain to be resolved, it is clear that GATE analysis identifies disease-relevant genes that include at least some promising therapeutic targets, in contrast with conventional SNP-by-SNP analysis of a GWAS, which identifies mostly SNPs of tiny effect near genes that often have no obvious relevance to disease-specific pathways. Because GATE scores typically account for only a small proportion of the variance of gene expression, they are unlikely to be useful for stratifying disease with respect to prognosis and prediction of drug response. The proteins and transcripts encoded by the genes that are identified by GATE analysis as core genes have much stronger associations with disease; establishing whether they are useful as clinical predictors will require validation studies in cohorts.
AUTHOR CONTRIBUTIONS
All authors contributed to at least one of the following manuscript preparation roles: conceptualization AND/OR methodology, software, investigation, formal analysis, data curation, visualization, and validation AND drafting or reviewing/editing the final draft. As corresponding author, Dr McKeigue confirms that all authors have provided the final approval of the version to be published and takes responsibility for the affirmations regarding article submission (eg, not under consideration by another journal), the integrity of the data presented, and the statements regarding compliance with institutional review board/Declaration of Helsinki requirements.
REFERENCES
1. McKeigue P. Quantifying performance of a diagnostic test as the expected information for discrimination: relation to the C-statistic. Stat Methods Med Res 2019; 28(6): 1841–1851.
2. Kuo C-F, Grainge MJ, Valdes AM, et al. Familial aggregation of rheumatoid arthritis and co-aggregation of autoimmune diseases in affected families: a nationwide population-based study. Rheumatology (Oxford) 2017; 56(6): 928–933.
3. Schaid DJ, Lin WY. One- and two-locus models for mapping rheumatoid arthritis-susceptibility genes on chromosome 6. BMC Proc 2007; 1(Suppl 1): S103.
4. Ishigaki K, Sakaue S, Terao C, et al; BioBank Japan Project. Multi-ancestry genome-wide association analyses identify novel genetic mechanisms in rheumatoid arthritis. Nat Genet 2022; 54(11): 1640–1651.
5. Fang H, Chen L, Knight JC. From genome-wide association studies to rational drug target prioritisation in inflammatory arthritis. Lancet Rheumatol 2020; 2(1): e50–e62.
6. Liu X, Li YI, Pritchard JK. Trans effects on gene expression can drive omnigenic inheritance. Cell 2019; 177(4): 1022–1034.e6.
7. Boyle EA, Li YI, Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic. Cell 2017; 169(7): 1177–1186.
8. Iakovliev A, McGurnaghan SJ, Hayward C, et al. Genome-wide aggregated trans-effects on risk of type 1 diabetes: a test of the “omnigenic” sparse effector hypothesis of complex trait genetics. Am J Hum Genet 2023; 110(6): 913–926.
9. Võsa U, Claringbould A, Westra HJ, et al; BIOS Consortium; i2QTL Consortium. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet 2021; 53(9): 1300–1310.
10. Sun BB, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature 2018; 558(7708): 73–79.
11. Ferkingstad E, Sulem P, Atlason BA, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet 2021; 53(12): 1712–1721.
12. Sun BB, Chiou J, Traylor M, et al; Alnylam Human Genetics; AstraZeneca Genomics Initiative; Biogen Biobank Team; Bristol Myers Squibb; Genentech Human Genetics; GlaxoSmithKline Genomic Sciences; Pfizer Integrative Biology; Population Analytics of Janssen Data Sciences; Regeneron Genetics Center. Plasma proteomic associations with genetics and health in the UK Biobank. Nature 2023; 622(7982): 329–338.
13. Fehrmann RSN, Jansen RC, Veldink JH, et al. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet 2011; 7(8):e1002197.
14. Hill MO. Diversity and evenness: a unifying notation and its consequences. Ecology 1973; 54(2): 427–432.
15. McKeigue PM, Spiliopoulou A, Iakovliev A, et al. Inference of causal and pleiotropic effects with multiple weak genetic instruments: application to effect of adiponectin on type 2 diabetes. medRxiv Preprint posted online December 17, 2023. doi:https://doi.org/10.1101/ 2023.12.15.23 300008
16. Fauman EB, Hyde C. An optimal variant to gene distance window derived from an empirical definition of cis and trans protein QTLs. BMC Bioinformatics 2022; 23(1): 169.
17. Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet 2001; 29(1): 19–20.
18. Diogo D, Kurreeman F, Stahl EA, et al; Consortium of Rheumatology Researchers of North America; Rheumatoid Arthritis Consortium International. Rare, low-frequency, and common variants in the protein-coding sequence of biological candidate genes from GWASs contribute to risk of rheumatoid arthritis. Am J Hum Genet 2013; 92(1): 15–27.
19. Zhou Q, Wang H, Schwartz DM, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haplo- insufficiency cause an early-onset autoinflammatory disease. Nat Genet 2016; 48(1): 67–73.
20. McGonagle D, Watad A, Savic S. Mechanistic immunological based classification of rheumatoid arthritis. Autoimmun Rev 2018; 17(11): 1115– 1123.
21. La Bella S, Rinaldi M, Di Ludovico A, et al. Genetic background and molecular mechanisms of juvenile idiopathic arthritis. Int J Mol Sci 2023; 24(3): 1846.
22. Orrù V, Steri M, Sole G, et al. Genetic variants regulating immune cell levels in health and disease. Cell 2013; 155(1): 242–256.
23. Tasaki S, Suzuki K, Kassai Y, et al. Multi-omics monitoring of drug response in rheumatoid arthritis in pursuit of molecular remission. Nat Commun 2018; 9(1): 2755.
24. Ren S, Tian Q, Amar N, et al. The immune checkpoint, HVEM
may contribute to immune escape in non-small cell lung cancer lacking PD-L1 expression. Lung Cancer 2018; 125: 115–120.
25.Ruth JH, Gurrea-Rubio M, Athukorala KS, et al. CD6 is a target for cancer immunotherapy. JCI Insight 2021; 6:e145662.
26. Helou DG, Shafiei-Jahani P, Hurrell BP, et al. LAIR-1 acts as an immune checkpoint on activated ILC2s and regulates the induction of airway hyperreactivity. J Allergy Clin Immunol 2022; 149(1): 223–236. e6.
27. Alotaibi FM, Min WP, Koropatnick J. CD5 blockade, a novel immune checkpoint inhibitor, enhances T cell anti-tumour immunity and delays tumour growth in mice harbouring poorly immunogenic 4T1 breast tumour homografts. Front Immunol 2024; 15:1256766.
28. Tiberio L, Laffranchi M, Zucchi G, et al. Inhibitory receptors of plasmacytoid dendritic cells as possible targets for checkpoint blockade in cancer. Front Immunol 2024; 15:1360291.
29. Henningsson L, Eneljung T, Jirholt P, et al. Disease-dependent local IL-10 production ameliorates collagen induced arthritis in mice. PLoS One 2012; 7(11):e49731.
30. Miura Y, Isogai S, Maeda S, et al. CTLA-4-Ig internalizes CD80 in fibroblast-like synoviocytes from chronic inflammatory arthritis mouse model. Sci Rep 2022; 12(1): 16363.
31. Plater-Zyberk C, Taylor PC, Blaylock MG, et al. Anti-CD5 therapy decreases severity of established disease in collagen type II-induced arthritis in DBA/1 mice. Clin Exp Immunol 1994; 98(3): 442–447.
32. Simelyte E, Rosengren S, Boyle DL, et al. Regulation of arthritis by p53: critical role of adaptive immunity. Arthritis Rheum 2005; 52(6): 1876–1884.
33. Zhao W, Dong Y, Wu C, et al. TIGIT overexpression diminishes the function of CD4 T cells and ameliorates the severity of rheumatoid arthritis in mouse models. Exp Cell Res 2016; 340(1): 132–138.
34. Pierer M, Schulz A, Rossol M, et al. Herpesvirus entry mediator-Ig treatment during immunization aggravates rheumatoid arthritis in the collagen-induced arthritis model. J Immunol 2009; 182: 3139–3145.
35. Kim S, Easterling ER, Price LC, et al. The role of leukocyte associated immunoglobulin-like receptor-1 (LAIR-1) in suppressing collagen-induced arthritis. J Immunol 2017; 199: 2692–2700.
36. Kwak HB, Ha H, Kim HN, et al. Reciprocal cross-talk between RANKL and interferon-gamma-inducible protein 10 is responsible for bone-erosive experimental arthritis. Arthritis Rheum 2008; 58(5): 1332–1342.
37. Boff D, Crijns H, Janssens R, et al. The chemokine fragment CXCL9 (74-103) diminishes neutrophil recruitment and joint inflammation in antigen-induced arthritis. J Leukoc Biol 2018; 104(2): 413– 422.
38. Wood MK, Daoud A, Talor MV, et al. Programmed death ligand 1-expressing macrophages and their protective role in the joint during arthritis. Arthritis Rheumatol 2024; 76(4): 553–565.
39. Maxwell L, Singh JA. Abatacept for rheumatoid arthritis. Cochrane Database Syst Rev 2009; 2009(4): CD007277.
40. Tuttle J, Drescher E, Simón-Campos JA, et al. A phase 2 trial of peresolimab for adults with rheumatoid arthritis. N Engl J Med 2023; 388(20): 1853–1862.
41. Zhai L, Ladomersky E, Lenzen A, et al. IDO1 in cancer: a Gemini of immune checkpoints. Cell Mol Immunol 2018; 15(5): 447–457.
42. Kagohashi K, Sasaki Y, Ozawa K, et al. Role of signal-transducing adaptor protein-1 for T cell activation and pathogenesis of autoimmune demyelination and airway inflammation. J Immunol 2024; 212: 951–961.
43. Sarangi PP, Chakraborty P, Dash SP, et al. Cell adhesion protein fibulin-7 and its C-terminal fragment negatively regulate monocyte and macrophage migration and functions in vitro and in vivo. FASEB J 2018; 32(9): 4889–4898.
44. Seki M, Oomizu S, Sakata KM, et al. Galectin-9 suppresses the generation of Th17, promotes the induction of regulatory T cells, and regulates experimental autoimmune arthritis. Clin Immunol 2008; 127(1): 78–88.
45. Sakaguchi S, Sakaguchi N, Asano M, et al. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (Cd25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 1995; 155: 1151–1164.
46. Seo SK, Choi JH, Kim YH, et al. 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med 2004; 10(10): 1088–1094.
47. Oflazoglu E, Boursalian TE, Zeng W, et al. Blocking of CD27-CD70 pathway by anti-CD70 antibody ameliorates joint disease in murine collagen-induced arthritis. J Immunol 2009; 183: 3770–3777.
48. Nozaki Y, Akiba H, Akazawa H, et al. Inhibition of the TIM-1 and -3 signaling pathway ameliorates disease in a murine model of rheumatoid arthritis. Clin Exp Immunol 2024; 218(1): 55–64.
49. Gu D, Ao X, Yang Y, et al. Soluble immune checkpoints in cancer: production, function and biological significance. J Immunother Cancer 2018; 6(1): 132.
50. Khan M, Zhao Z, Arooj S, et al. Soluble PD-1: predictive, prognostic, and therapeutic value for cancer immunotherapy. Front Immunol 2020; 11:587460.
51. Wang X, Goldstein DB. Enhancer domains predict gene pathogenicity and inform gene discovery in complex disease. Am J Hum Genet 2020; 106(2): 215–233.
52. Mostafavi H, Spence JP, Naqvi S, et al. Systematic differences in discovery of genetic effects on gene expression and complex traits. Nat Genet 2023; 55(11): 1866–1875.
Credits: Spiliopoulou A, Iakovliev A, Plant D, Sutcliffe M, Sharma S, Cubuk C, Lewis M, Pitzalis C, Barton A, McKeigue PM. Genome-Wide Aggregated Trans Effects Analysis Identifies Genes Encoding Immune Checkpoints as Core Genes for Rheumatoid Arthritis. Arthritis Rheumatol. 2025 Jul;77(7):817-826. doi: 10.1002/ art.43125. Epub 2025 Mar 16. PMID: 39887658; PMCID: PMC12209750.






