

Vivien Li, Baptiste Leurent, Frederik Barkhof, Marie Braisher, Fay Cafferty, Olga Ciccarelli, Arman Eshaghi, Emma Gray, Jennifer M. Nicholas, Mahesh Parmar, Guy Peryer, Jenny Robertson, Nigel Stallard, James Wason, Jeremy Chataway
Affiliation
1 From the Florey Institute of Neuroscience and Mental Health (V.L.), University of Melbourne; Department of Neurology (V.L.), Royal Melbourne Hospital, Australia; Department of Medical Statistics (B.L., J.M.N.) and International Statistics and Epidemiology Group (B.L.), London School of Hygiene and Tropical Medicine, UK; Department of Radiology and Nuclear Medicine, Neuroscience Campus Amsterdam (F.B.), VU University Medical Center, Amsterdam, the Netherlands; Queen Square Institute of Neurology and Centre for Medical Image Computing (F.B.), Department of Neuroinflammation, Queen Square Multiple Sclerosis Centre (M.B., O.C.), and NMR Unit, Department of Neuroinflammation (A.E.), Faculty of Brain Sciences, UCL Queen Square Institute of Neurology; MRC Clinical Trials Unit at UCL, Institute of Clinical Trials and Methodology (F.C., M.P., J.C.), and Department of Computer Science, Centre for Medical Image Computing (A.E.), University College London; National Institute for Health Research (F.B., O.C., J.C.), University College London Hospitals Biomedical Research Centre; UK Multiple Sclerosis Society (E.G., G.P., J.R.), London; Faculty of Medicine and Health Sciences (G.P.), University of East Anglia, Norwich; Statistics and Epidemiology, Division of Health Sciences (N.S.), Warwick Medical School, University of Warwick, Coventry; and Population Health Sciences Institute (J.W.), Newcastle University, UK.
Abstract
There are few treatments shown to slow disability progression in progressive multiple sclerosis (PMS). One challenge has been efficiently testing the pipeline of candidate therapies from preclinical studies into clinical trials. Multi-arm multi-stage (MAMS) platform trials may accelerate the evaluation of new therapies compared to traditional sequential clinical trials. We describe a MAMS design in PMS focusing on the selection of interim and final outcome measures, sample size, and statistical considerations. The UK MS Society Expert Consortium for Progression in MS Clinical Trials reviewed recent phase II and III PMS trials to inform interim and final outcome selection and design measures. Simulations were performed to evaluate trial operating characteristics under different treatment effects, recruitment rate, and sample size assumptions. People with MS formed a patient and public involvement group and contributed to the trial design, ensuring it would meet the needs of the MS community. The proposed design evaluates 3 experimental arms compared to a common standard of care arm in 2 stages. Stage 1 (interim) outcome will be whole-brain atrophy on MRI at 18 months, assessed for 123 participants per arm. Treatments with sufficient evidence for slowing brain atrophy will continue to the second stage. The stage 2 (final) outcome will be time to 6-month confirmed disability progression, based on a composite clinical score comprising the Expanded Disability Status Scale, Timed 25-Foot Walk test, and 9-Hole Peg Test. To detect a hazard ratio of 0.75 for this primary final outcome with 90% power, 600 participants per arm are required. Assuming one treatment progresses to stage 2, the trial will recruit ≈1,900 participants and last ≈6 years. This is approximately two-thirds the size and half the time of separate 2-arm phase II and III trials. The proposed MAMS trial design will substantially reduce duration and sample size compared to traditional clinical trials, accelerating the discovery of effective treatments for PMS. The design was well-received by people with multiple sclerosis. The practical and statistical principles of MAMS trial design may be applicable to other neurodegenerative conditions to facilitate efficient testing of new therapies.
Glossary
9HPT = 9-Hole Peg Test; EDSS = Expanded Disability Status Scale; MAMS = multi-arm multistage; MS = multiple sclerosis; PBVC = percentage of brain volume change; PMS = progressive multiple sclerosis; PPI = patient and public involvement; PPMS = primary progressive multiple sclerosis; RRMS = relapsing-remitting multiple sclerosis; SPMS = secondary progressive multiple sclerosis; T25FW = Timed 25-Foot Walk
Progressive multiple sclerosis (PMS) is a significant health problem worldwide1 and has considerable financial costs for health care systems, patients, and their caregivers, with costs increasing at higher levels of disability.2,4 Despite extensive efforts, there are few proven therapies for PMS. Compared to the predominantly inflammatory pathology in relapsing multiple sclerosis (MS) targeted by current treatments, the neurodegenerative processes driving progression in PMS are complex and less well-defined. 5,6 There is a pipeline of candidate therapies from preclinical studies, but the challenge is testing them efficiently in clinical trials with appropriate outcome measures to determine whether they can successfully slow disability progression.
One potential avenue is improving the efficiency of trials by incorporating adaptive elements in a multi-arm multistage (MAMS) platform design. MAMS trials aim to evaluate multiple experimental arms and seamlessly integrate traditional phase II and III evaluations into a single trial. They have been successful in accelerating the evaluation of therapies and changing practice in other disease settings, such as cancer7 and infectious diseases.8 They are also increasingly being considered for neurologic conditions such as Alzheimer’s disease,9 Parkinson’s disease10 and motor neuron disease.11,12 These neurodegenerative conditions share commonalities with PMS, where there is a marked translational gap between the relative abundance of early phase trials stemming from increased understanding of disease pathobiology and the lack of positive phase III trials leading to disease-modifying treatments.
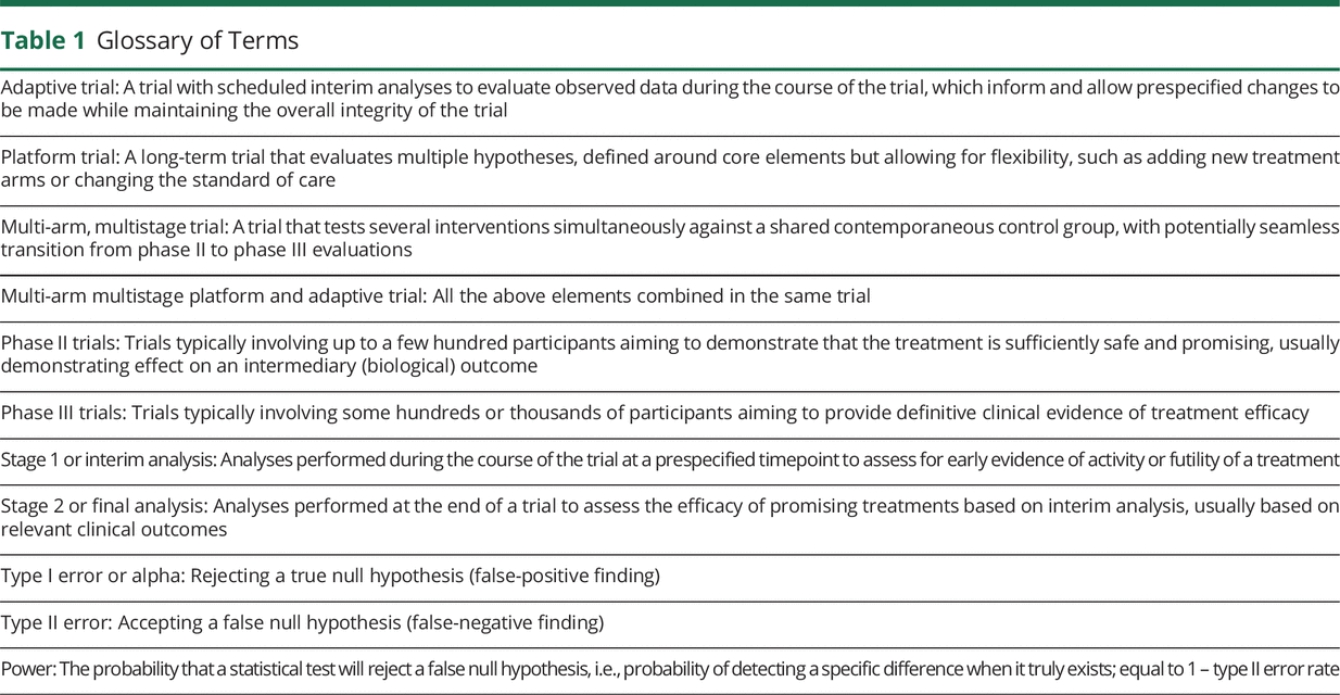
Adaptive MAMS platform designs offer flexible features that can provide efficiencies at various levels13 (Table 1). These include simultaneous evaluation of multiple treatments against a common standard of care, reducing both time and numbers of patients required; the ability to add new treatments as they become relevant, avoiding lengthy setup times for multiple trials; dropping treatments that are not showing sufficient promise, allowing redirection of resources; and incorporation of the traditional separate phase II and III evaluations within a single protocol with seamless transitions.
With the aim of designing a MAMS trial in PMS, the UK MS Society Expert Consortium for Progression in MS Clinical Trials set up 4 working groups on outcome measures, trial design, treatment selection, and trial infrastructure. Each group included members with relevant expertise and worked closely with the patient and public involvement (PPI) group throughout the development process. The work of the treatment selection working group on identifying and shortlisting candidate treatments, focusing on licensed drugs that can be repurposed, has been reported elsewhere.14
This article describes the work of the trial design and outcome measures working groups. We discuss key elements of the MAMS trial design based on the evaluation of 3 candidate treatments against the standard of care in 2 analysis stages, including the selection of the primary interim and final outcomes, sample size, and other statistical considerations.
Methods
Outcome Measures
The outcome measures a working group comprised of individuals with expertise in MS trials, imaging, and biomarkers, as well as people with MS with lived experience of the condition. The group reviewed the literature to determine outcome measures relevant to a MAMS trial evaluating predominantly neuroprotective treatments. Individual members submitted proposed outcome measures based on their expertise with the final prioritization of outcomes determined in consensus meetings.
The MAMS design allows distinct interim (stage 1) and final (stage 2) outcomes. The final outcome should be clinically derived and relevant to patients and regulators. The interim outcome serves as an early indicator of whether a treatment is likely to be effective and hence should be continued into the second stage of the trial while minimizing the likelihood of ceasing truly effective treatments. It should reflect the underlying association between the treatment and the clinical outcome. The absence of effect on the interim outcome should be indicative of the absence of effect on the final outcome, although the converse may not necessarily hold.15
Trial Design
The trial design working group, comprising experts in the design and implementation of MAMS trials, statisticians, and MS clinicians, was tasked with generating design options for running an efficient, scalable, and flexible clinical trial by exploring different scenarios to determine the best design type. The group reviewed data from phase II and III randomized controlled PMS trials from January 1, 2009, to January 1, 2019, to inform key design measures for both stage 1 and 2 analyses, such as effect size, and the relationship between the interim and final outcomes.
To assess the statistical operating characteristics of the trial (e.g., type I and type II error rates), we simulated multiple trials with different correlation structures for the treatment effects on stage 1 and stage 2 outcomes, under different treatment effect assumptions. We also modelled the expected trial progress over time based on different assumptions (such as recruitment rates) and design parameters (such as sample size and treatment stopping rules) to the second stage. Further details of the simulation methods are reported in eAppendix 1 and 2 (links. lww.com/WNL/B900). To support the design of the stage 1 analysis, we analyzed brain atrophy data from the MS-STAT116 and ASCEND17 clinical trials (for full Methods, see eAppendix 1, links.lww.com/WNL/B900). Modelling was conducted in Stata version 15 (StataCorp) and Microsoft Excel 2016.
Patient and Public Involvement
A PPI strategy group was involved since the earliest conception of the project, including members of each of the expert consortium working groups. The PPI strategy group included 4 members of the MS Society Research Network (patients with MS) and the MS Society Public Involvement Officer. They contributed to discussions as the project developed and focused on ensuring that the research would meet the needs of the MS community. Additional workshops attended by a total of 34 people with MS held in 3 UK locations brought in further expertise of people with MS on topics including relevance, feasibility, and acceptability of all aspects of the trial design as well as recruitment and engagement strategies.
Data Availability
Data not published in this article will be made available by request from any qualified investigator.
Review of Previous Trials
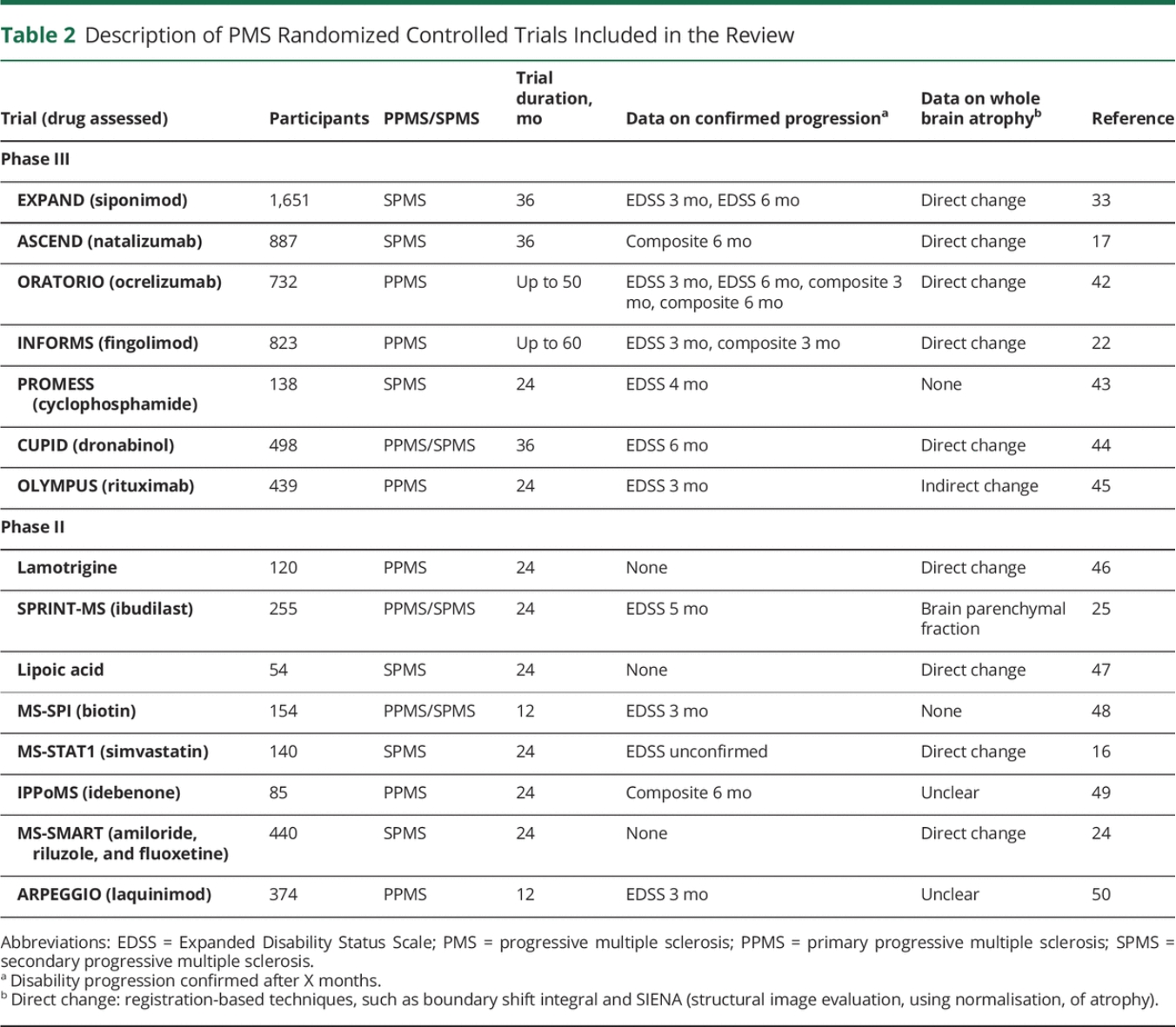
Our review identified 15 eligible phase II (n = 8) and phase III (n = 7) randomized trials in PMS (Table 2). The median trial size was 374 participants (range 54–1,651) and the median follow-up duration was 2 years (range 1–4.5 years). Trials included secondary progressive MS (SPMS) (n = 6), primary progressive MS (PPMS) (n = 6), and mixed PMS (n = 3). Confirmed disability progression on the Expanded Disability Status Scale (EDSS) was reported in 9 trials at different time intervals ranging from 3 to 6 months and a composite outcome was reported in 4. Two (29%) of the phase III trials (siponimod, ocrelizumab) and 4 (50%) of the phase II trials (ibudilast, lipoic acid, biotin, simvastatin) were positive for their primary endpoints.
Proposed MAMS Trial Design
Overview
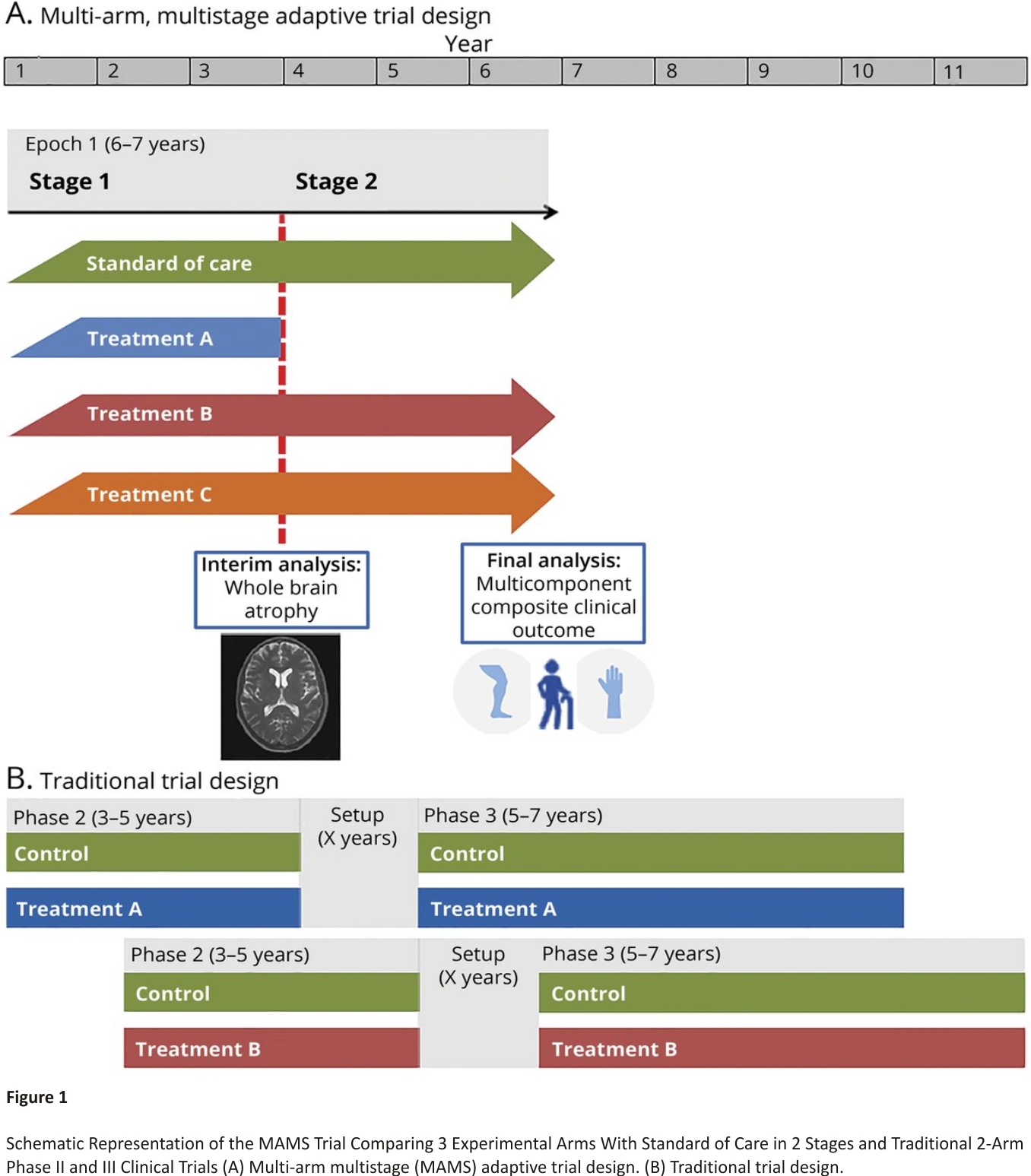
We propose the design of a 2-stage MAMS trial in PMS: 1 interim analysis to examine early evidence of treatment effect (stage 1) and 1 final confirmatory analysis of efficacy (stage 2). The trial would include 4 arms in stage 1: 1 standard of care (control) arm and 3 experimental arms. Any treatment that is sufficiently promising at the interim analysis will progress to stage 2, which will continue until the required number of events is reached, as represented in Figure 1.
It is expected that in the initial phase of the trial the standard of care arm for most participants will comprise the best supportive care. Whereas ocrelizumab and siponimod have been approved for PMS, these treatments are not currently available to or suitable for all patients, in particular nonambulatory patients, who would be eligible for this proposed trial. If an efficacious therapy is subsequently found, this would then become the standard of care for future participants entering the platform.
The number of experimental arms was informed by feasibility constraints and the treatment selection group’s work on a number of repurposed therapies ready for clinical testing.14 Participants will be randomized with an equal probability between each of the 4 arms (1:1:1:1 ratio). In a standard multi-arm trial with n experimental arms, the optimal allocation ratio would be n: 1 in favour of the control arm. This is because the control participants contribute to each of the pairwise comparisons. However, for a MAMS trial, this depends on the number of arms continuing into stage 2, which is unknown.18 Unequal allocations would also make the trial less attractive to people with MS, as it results in a lower likelihood of being randomized to an experimental arm.
Choice of Stage 2 (Final) Primary Outcome
The classic measurement tool and the regulatory standard has been the EDSS,19 used to determine the time to disability progression. Its strengths and limitations are well-documented20 and numerous attempts have been made to evolve it, including using a composite measure based on progression in 1 or more of 3 endpoints: (1) increase in EDSS (of ≥1 point if baseline EDSS was <5.5 or ≥0.5 points if baseline EDSS was ≥5.5), (2) ≥20% increase in 9-Hole Peg Test (9HPT), or (3) ≥20% increase in Timed 25-Foot Walk (T25FW) (if ambulant).21
Composite measures achieve higher event rates than single measures (eAppendix 3, links.lww.com/WNL/ B900), which can reduce the trial duration and sample size. For example, in the INFORMS phase III trial of fingolimod in PPMS, >70% of participants had reached progression on the 3-month confirmed disability composite outcome by 3 years, as opposed to 50% based on EDSS alone.22 Inclusion of a measure of upper limb function also addresses the PPI group’s interest in expanding the traditionally narrow EDSS inclusion criteria to include patients with higher levels of disability, to whom arm function is critical and measures of ambulation less relevant.23
Based on these considerations, we selected time to 6-month confirmed composite disability progression as the primary outcome for the final (stage 2) analysis. The composite outcome will be measured at baseline and every 6 months until the end of the follow-up. The time to progression will be from randomization until the date of the initial disability progression (if subsequently confirmed). Based on earlier trials (eAppendix 3, links.lww. com/WNL/B900), we expect the rate of 6-month confirmed composite disability progression to be around 50% at 3 years.
Choice of Stage 1 (Interim) Primary Outcome
Whole-brain atrophy on MRI, measured as an annualized percentage of brain volume change (PBVC), was selected as the primary interim outcome, based on the initial candidate drugs having primarily neuroprotective mechanisms of action. Brain atrophy reflects underlying neuroaxonal loss, which contributes to the accrual of disability in PMS, and has been successfully used as a primary outcome in phase II trials, including MS-STAT1,16 MS-SMART,24 and SPRINT-MS.25 Importantly for a multistage trial, the treatment effect size on atrophy has been found to correlate with the clinical disability endpoint in a meta-analysis of relapsing-remitting MS (RRMS) trials.24
Methods of measuring PBVC include registration-based techniques (such as boundary shift integral and SIENA [structural image evaluation, using normalisation, of atrophy]) or brain parenchymal fraction, which quantify the amount of brain tissue contained within a contour surrounding the entire brain including CSF.26 Some therapies, particularly those with anti-inflammatory effects, can excessively reduce brain volume in the first months (pseudoatrophy),27 so it was recommended to assess PBVC also after at least 6 months on treatment.
We considered other imaging-based measures, including spinal cord atrophy, which contributes to MS disability progression and occurs at a faster rate than brain atrophy,28 neurite indices derived from diffusion MRI, which reflect the microstructural changes of axons and dendrites, and magnetization transfer imaging, which reflects demyelination and axonal loss.19 However, technical challenges limit widespread implementation and standardization across multiple centres.27 Although biofluid markers such as neurofilament light chain are associated in high concentrations with disability and brain atrophy,29 there are mixed findings on whether they are sensitive to treatment and they are not ready to be used as primary outcome measures until a validated, standardized, and widely accessible assay is available, with normative values of neurofilaments across age groups. Moreover, there is divergence of their utility in relapsing MS compared to PMS.30,31
Predicted Brain Atrophy Rate
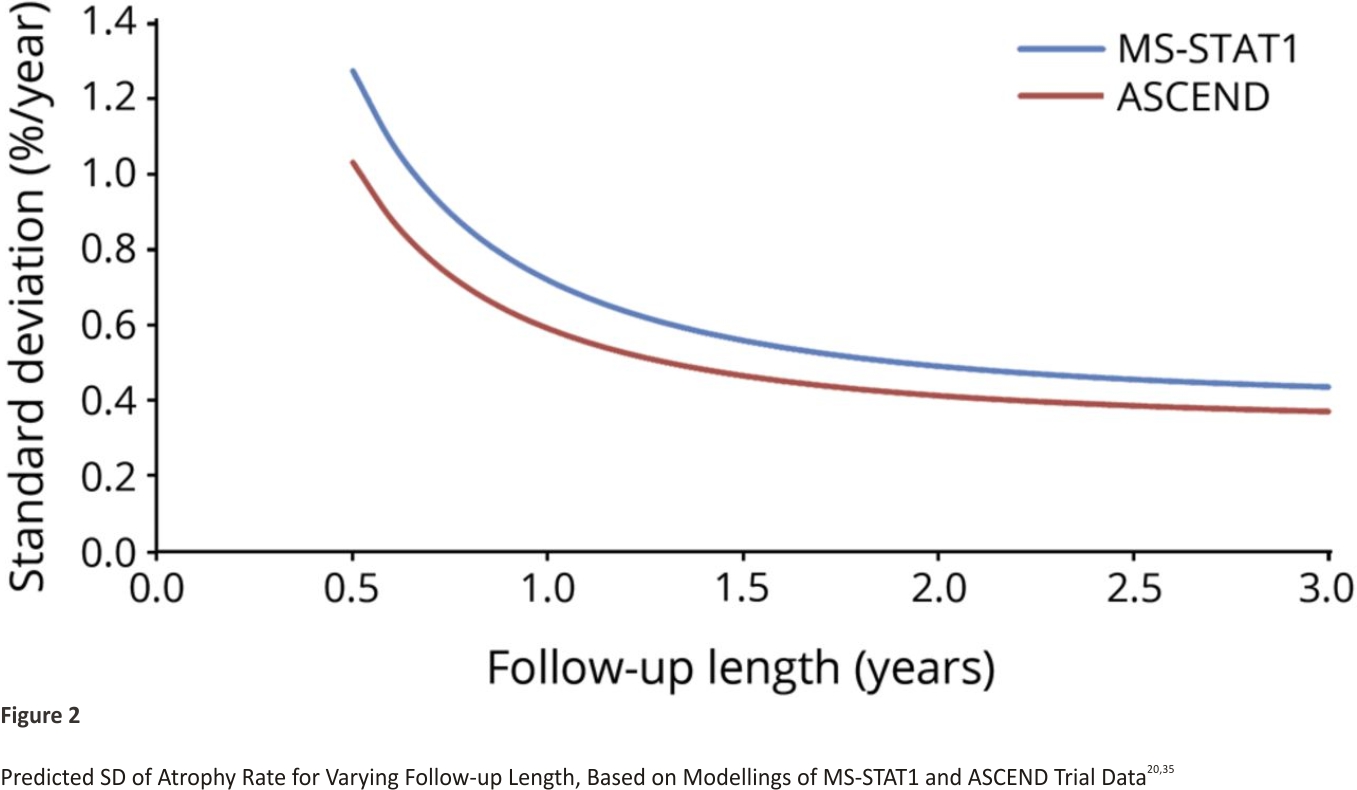
Nine of the reviewed trials reported a direct measure of change in whole brain volume (eAppendix 1, links.lww. com/WNL/B900). Brain atrophy rates varied between 0.4%/year and 0.7%/ year in control arms. There was no clear pattern of differences between trials in PPMS or SPMS or by follow-up length. The SD for atrophy rate decreased with increasing follow-up length, ranging from 0.59%/year to 0.78%/year over 1 year, and 0.37%/ year to 0.60%/year over 2 years.
The predicted SDs based on applying our statistical model (eAppendix 1, links.lww.com/WNL/B900) to the data from MS-STAT1 and ASCEND is shown in Figure 2. The SD is expected to decrease rapidly with the increasing length of follow-up, especially in the first 12 months. After 18–24 months, the reduction in SD becomes much smaller.
The timing of the interim analysis is an important consideration in adaptive designs. It should occur after accruing sufficient participant data to make a reliable decision on continuing or dropping treatment arms, but early enough relative to total trial recruitment to have value in informing adaptation of the trial design.32 Based on these considerations, PBVC after 18 months of follow-up will be assessed at interim analysis. This choice achieves a balance between reducing the variance of the measure and ensuring that the interim analysis was sufficiently timely to make it worthwhile (see below). The SD at this point is predicted to be around 0.55%/year.
Treatment Effects on Brain Atrophy and Clinical Progression
A key criterion for the stage 1 outcome is the ability to identify treatments expected to be ineffective and also potentially effective in terms of the final (stage 2) outcome. We reviewed trials reporting treatment effects on both brain atrophy rate and clinical progression. Trial results are reported in eAppendix 2 (links.lww.com/WNL/ B900) and summarized in Figure 3. There is a negative correlation, indicating that drugs with a stronger effect on reducing brain atrophy in PMS were more effective on clinical outcomes, confirming findings in RRMS.26
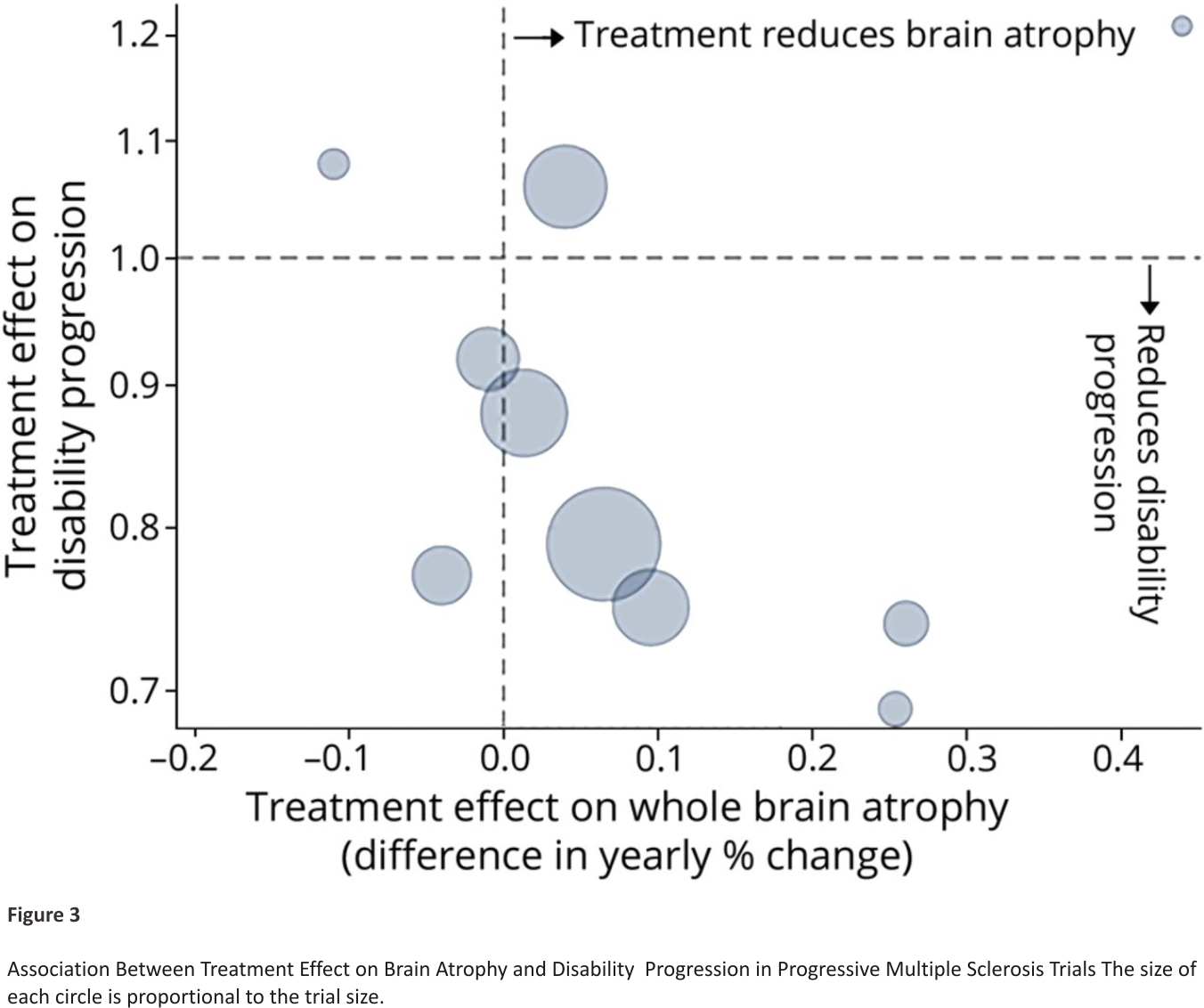
Our trial targets a treatment effect of 25% relative reduction in the 6-month confirmed disability progression rate, that is, a hazard ratio of 0.75. This is a clinically important effect in slowing progression in ambulation, upper limb function, or disability, which has been achieved in previous trials.33 Assuming 50% of patients experience a disability progression by 3 years in the control arm, a 25% relative reduction would equate to a 12.5% absolute difference (50% control vs 37.5% active treatment).
Based on the review of previous trials, we assumed an effective treatment would reduce the rate of whole-brain atrophy by around 0.15%/ year, from 0.55%/year to 0.40%/year.
Stage 1 Sample Size
The sample size for stage 1 analysis was based on pairwise comparisons of whole-brain atrophy rate at 18 months between each intervention arm and standard of care. A 1-sided test is used for the interim analysis, with treatment continuing to the second stage if there is evidence in favour of a lower atrophy rate compared to standard of care. We chose 95% power because a priority of the interim analysis is to minimize the chance of stopping an arm when the treatment is genuinely active in slowing brain atrophy (i.e., avoid false negatives). Stage 1 alpha (type I error rate) captures the probability of an ineffective treatment to continue to the second stage. It should be chosen to balance minimizing this risk while ensuring the timeliness of the interim analysis. Designs were considered with stage 1 alpha between 20% and 50% with the final choice of 35% representing an achievable sample size and timely interim analysis (see trial timeline). This is in line with other MAMS trials,15 but differs from the 5% commonly used in the confirmatory analysis, as the objectives here are different. Assuming an SD of 0.55%/year (see above), 111 observations per arm will allow 95% power to detect a 0.15%/year difference at a 1-sided significance level (alpha) of 35%. Allowing for 10% drop-out, 123 participants are needed per arm.
Therefore, we recommended that stage 1 analysis be conducted once 18 months of brain atrophy data are available for 111 participants per arm, with pairwise comparison for each experimental arm compared to the control arm. If the 1-sided p-value is below 0.35, then the treatment arm is continued into stage 2.
Stage 2 Sample Size
The sample size for the stage 2 analysis was based on comparing the time to confirm disability progression between each intervention arm to the standard of care. For each pairwise comparison, to have 90% stage 2 power to detect a hazard ratio of 0.75 at the 2-sided stage 2 significance level of 5% (or equivalently a 2.5% 1-sided significance level), 281 progression events are required in the control arm and 600 participants per arm are needed.
The stage 2 significance level was set at a 2-sided 5% level, as in standard confirmatory trials, corresponding to a 1-sided level of 0.025. The question of multiplicity (adjusting significance level due to multiple comparisons) has been discussed before in MAMS.18 We aimed to select drugs with different mechanisms, which might be viewed as independent evaluations, similar to multiple trials being conducted,34 and therefore did not apply any correction for multiple comparisons. If drugs of similar action are selected (e.g., different doses of the same drug), an appropriate correction (e.g., Dunnett35) should probably be applied. The statistical power in a time-to-event analysis is determined by the number of events. Recruiting 600 participants per arm should be sufficient to observe the required 281 progression events in the control arm in a timely manner. This number of events is anticipated to occur around 18 months after the last participant has been enrolled, assuming a 10% drop-out rate and 50% disability progression rate by 3 years and recruitment rate, as described in eAppendix 4 (links. lww.com/WNL/B900) (see trial timeline).
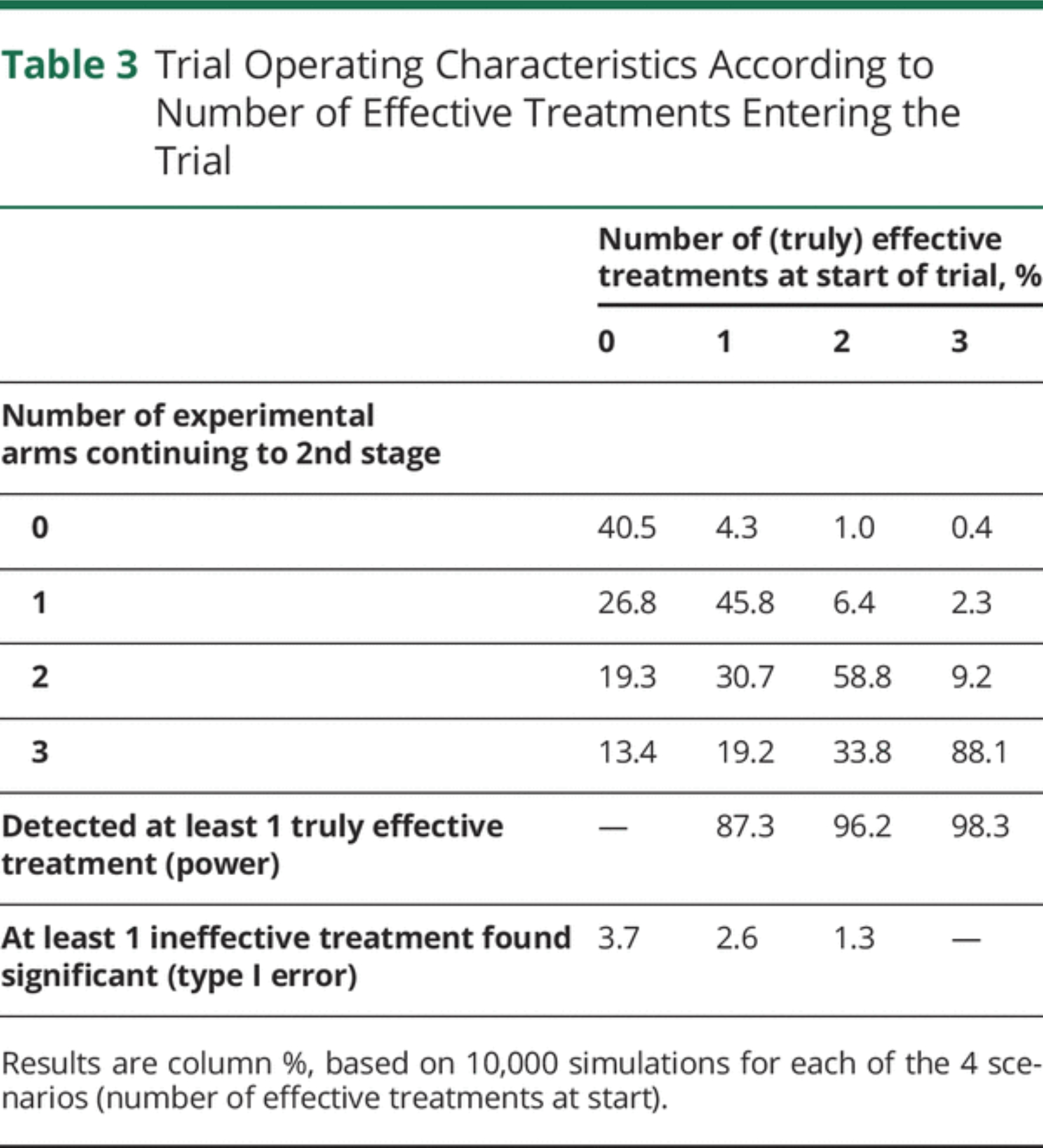
Trial Operating Characteristics
We conducted simulations to assess the operating characteristics of the proposed trial design under different scenarios (see eAppendix 5, links.lww. com/WNL/B900, for Methods and full Results). Table 3 shows the overall trial characteristics, depending on the number of truly effective treatments at the start of the trial. In all scenarios, the probability of wrongly concluding that 1 or more treatments are effective (false-positive) is below 4%. The chance of correctly concluding that at least 1 treatment is effective (power) if a single effective drug enters the trial is around 87%, but this increases to above 96% if more than 1 effective drug enters the trial.
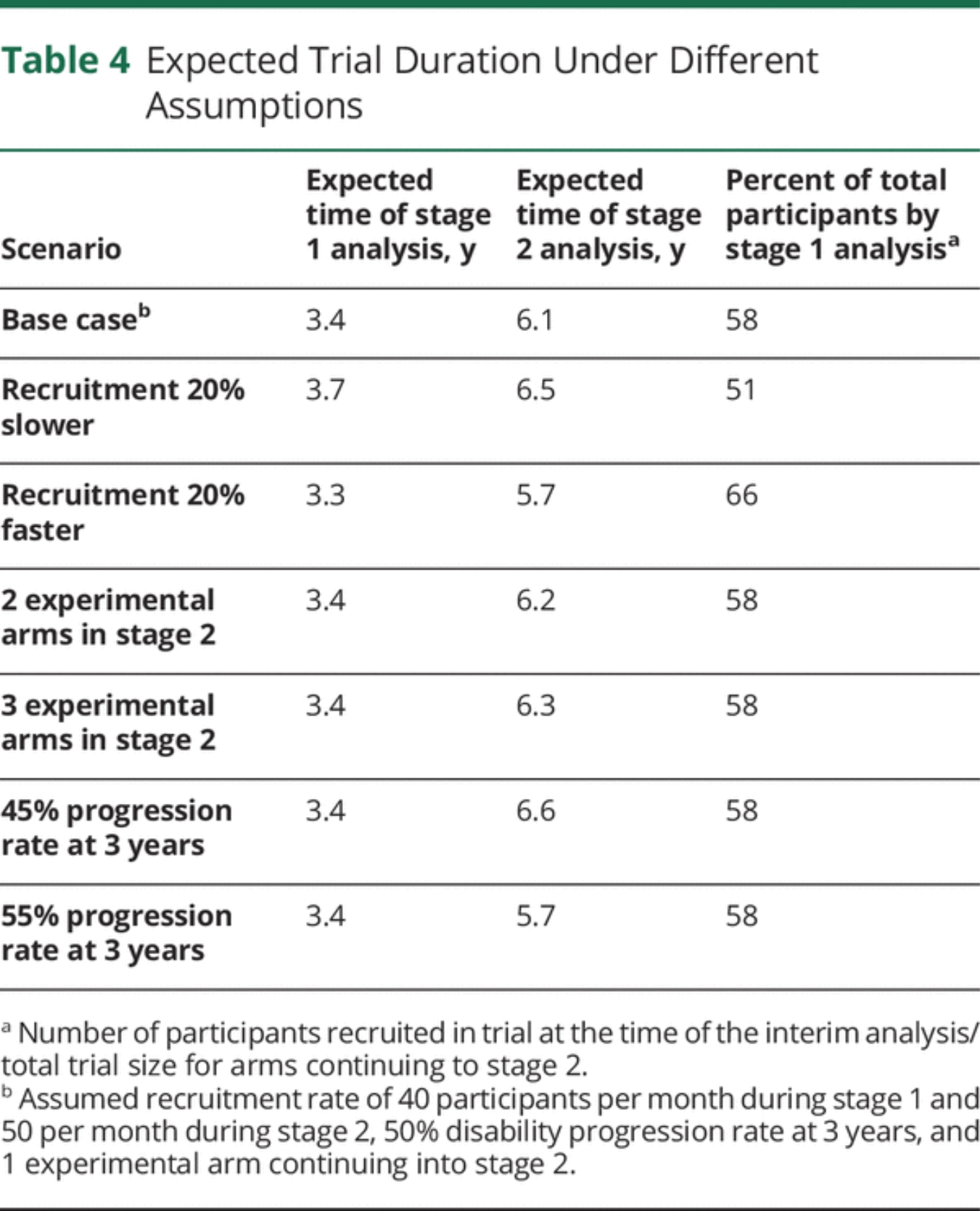
Trial Timeline
An important consideration in adaptive trials is to anticipate the possible dynamics of the trial over time, including the relative timing of the interim and final analyses. eAppendix 4 (links.lww.com/WNL/B900) describes the assumptions made and how the timeline was modelled. Results are summarized in Table 4. Under a base case scenario of 40–50 participants recruited per month and 1 experimental arm continuing into stage 2, we expect the interim analysis to be conducted after around 3.4 years, and the final analysis after 6.1 years (ranging between 5.7 and 6.6 years depending on different scenarios modelled).
Discussion
MAMS trials have considerable potential in PMS, where there are many candidate therapies, as well as relevant interim outcome measures that have an appropriate relationship to final clinical outcomes. We propose a MAMS trial design that could potentially accelerate the evaluation of new treatments in PMS.
Advantages
The proposed MAMS design leads to efficiencies in both sample size and trial duration compared to traditional separate phase II and III trials of single treatments. A single control arm is used to assess multiple experimental arms and participants recruited in stage 1 seamlessly continue to be included in the stage 2 analysis without additional setup time in between. This trial is expected to last 6–7 years with 1,900 participants, encompassing the stage 1 and 2 evaluations of 3 initial candidate treatments (Figure 1 and eAppendix 4, links.lww.com/WNL/B9 00).
In comparison, under the traditional approach, around 630 patients would be required in each of 3 phase II studies to have 90% power with 5% type I error under the same assumptions. If one of these treatments was found to be effective and proceeded to phase III, 1,200 additional participants would be required, totalling 3,090 participants. Separate phase II and III trials of a single treatment would be expected to take more than 10 years, with 3–5 years for phase II, 5–7 years for phase III, and additional setup time between the two (Figure 1). For example, the evaluation of high-dose simvastatin is following a more conventional path with separate phase II (MS-STAT)16 and phase III (MS-STAT2)36 trials. Recruitment to MS-STAT started in 2011 and MS-STAT2 is expected to be completed by 2025, which corresponds to 14 years overall.
Challenges
Planning and setting up a MAMS adaptive platform trial is considerably more complex than standard phase II and III trials and may take up to 12 to 18 months. In particular, statistical simulations examining different design options, scenarios, and parameters are essential to optimize efficiency and select appropriate trial operating characteristics while preserving the overall integrity of the trial. The initial modest investment in time and resources will be further offset by shorter subsequent setup times for further treatments added to the platform.
As adaptive platform designs are relatively novel in neurodegenerative diseases, there is a perception that regulatory agencies may not immediately accept them as equal to more conventional phase III studies. However, a precedent has been set for regulatory approval of MAMS platform trials in settings such as oncology7 and infectious diseases8 and our experience in these other disease areas suggests that regulators are becoming more open to, knowledgeable and welcoming of such designs.
Patient and Public Involvement
The PPI group actively participated in the entire trial design process, as well as treatment and outcome measure selection, to ensure the needs of people with MS were being met. For example, it was important, particularly for nonambulatory people with MS, to include an assessment of upper limb function in the primary efficacy endpoint and proposed secondary outcomes including patient-reported outcome measures of key symptoms such as fatigue. Feedback indicated the trial design was well-received and acceptable, despite being more complex. Perceived advantages included the ability to evaluate multiple candidate treatments and the relatively lower likelihood of randomization to placebo. If a participant’s treatment arm is discontinued after interim analysis, there is the potential opportunity to re-enter the trial in a continuing arm or future trials in the platform. Some participants expressed concern about the total trial duration, but this was offset by the favourable consensus overall regarding the potential acceleration of treatment discovery.
Scope and Future Developments
This article is based on work conducted by the trial design working group and in many senses is an evolution from our work carried out a decade ago.37 A program grant proposal based on the activity of all working groups of the UK MS Society’s Expert Consortium for Progression in MS was submitted to the UK MS Society in November 2019 and received favourable international peer and lay review. Funding has been awarded to develop the protocol and deliver the first active arms plus standard of care in the MAMS trial platform, with recruitment expected to commence in 2022. Whereas this article focuses on the evaluation of only the first 3 candidate therapies, the adaptive MAMS platform will allow the addition of new treatment arms7 and pre-randomization of participants from discontinued arms in the future.38 Drugs with predominantly remyelinating potential will likely require additional and alternative endpoints at the interim analysis stage. Further aspects of the trial protocol, for example, secondary and exploratory outcomes and recruitment infrastructure, are beyond the scope of this article.
Adaptive Platform Trials in Other Neurologic Disorders
Like PMS, conditions such as Parkinson’s disease, Alzheimer’s disease, and motor neuron disease are increasing in prevalence, have a significant impact on patients, carers, and health care systems, and have no or few therapies that slow or prevent progression. An improved understanding of disease pathophysiology in recent years has led to a growing pipeline of potential therapeutics. For example, a 2020 review identified 121 agents in 136 phase I to III clinical trials for Alzheimer’s disease, with an increasing number of disease modification treatment candidates over the past 5 years.39 However, these conditions face similar challenges in efficiently translating candidate drugs into effective treatments with many disappointing phase III clinical trial results to date. Various reasons for this have been proposed, including the need to improve the trial design.10,12,40
MAMS designs are particularly relevant when there are multiple candidate therapies to be trialled and when a reliable early marker of clinical efficacy is available. MAMS adaptive platform trials have been planned and initiated to accelerate successful drug discovery in these disorders. The Motor Neuron Disease–Systematic Multi-arm Adaptive Randomization Trial (MND-SMART) will initially test 2 repurposed drugs against a common placebo.11 The Dominantly Inherited Alzheimer’s Network Trials Unit (DIAN-TU) platform trial established in 2012 was a multi-arm trial of 2 anti-amyloid monoclonal antibodies. Although neither drug met the primary cognitive endpoint,41 lessons learned, including refinements in participant and outcome measure selection and trial duration, have led to several emerging platform trials, such as the AHEAD study evaluating different doses of an anti-Aβ monoclonal antibody in 2 phase III clinical trials that respectively use amyloid PET and cognitive testing as the primary outcome measures.9
The principles of designing a PMS MAMS trial as outlined in this article are relevant to other neurodegenerative conditions, but each condition will present unique considerations and challenges, including a selection of biologically and clinically relevant, sensitive, and timely interim and final outcome measures, determination of the most appropriate patient population for inclusion, and trial duration required to detect a meaningful effect.
We propose a design for a MAMS trial in PMS for the evaluation of 3 repurposed neuroprotective drugs compared to the standard of care. Although more complex in design, efficiencies in participant numbers and trial duration, as well as the ability to incorporate adaptive elements and continually test newly identified treatments through an ongoing platform, make this approach more likely to succeed in finding effective therapies that target disability progression in PMS in a timely manner.
Study Funding
This work was supported by UK Multiple Sclerosis Society project grants 101 and 110; an award from the International Progressive MS Alliance (reference number PA-1412-02,420); and the ACORD collaboration: A Collaboration of Groups Developing, Running and Reporting Multi-arm, Multi-stage Trials in Neurodegenerative Diseases (mrcctu.ucl.ac.uk/studies/ all-studies/a/acord/).
Disclosure
V. Li and B. Leurent report no disclosures relevant to the manuscript. F. Barkhof acts as a consultant for Bayer-Schering, Biogen-Idec, IXICO, Merck-Serono, Novartis, Janssen, and Roche; has received grants from IMI for the Amyloid Imaging to Prevent Alzheimer’s Disease (AMYPAD) initiative, the Dutch MS Society, ECTRIMS-MAGNIMS, the Dutch Research Council (NWO), the UK MS Society, Biogen, Merck, and GE Healthcare; is supported by the UCLH Biomedical Research Centre through the National Institute for Health Research (NIHR); is co-founder and a stockholder of Queen Square Analytics Ltd.; and is on the editorial boards of Radiology, Neuroradiology, Multiple Sclerosis Journal, and Neurology®. M. Braisher and F. Cafferty report no disclosures relevant to the manuscript. O. Ciccarelli is Deputy Editor of Neurology and serves on the editorial board of Multiple Sclerosis Journal; and receives research support from the NIHR UCLH/UCL Biomedical Research Centre, NIHR, MS Society of Great Britain and Northern Ireland, National MS Society, and Rosetrees Trust. A. Eshaghi has received speaker’s honoraria from Biogen and At The Limits educational programme; travel support from the National Multiple Sclerosis Society; honoraria from the Journal of Neurology, Neurosurgery and Psychiatry for editorial commentaries; research grants from Biogen and Roche through his institutions; holds an equity stake in Queen Square Analytics, and serves on the editorial board of Neurology. E. Gray, J. Nicholas, M. Parmar, G. Peryer, J. Robertson, N. Stallard, and J. Wason report no disclosures relevant to the manuscript. J. Chataway has received support from the Efficacy and Evaluation Programme, a Medical Research Council and NIHR partnership, the Health Technology Assessment Programme (NIHR), the UK MS Society, the US National MS Society, and the Rosetrees Trust in the past 3 years; and is supported in part by the National Institute for Health Research, University College London Hospitals, Biomedical Research Centre, London, UK. Go to Neurology.org/N for full disclosures.
Acknowledgement
Susan Scott, a Research Network member of the UK Multiple Sclerosis Society, provided advice during the trial design process and reviewed the manuscript.
References
1. International MS Federation. Atlas of MS, 3rd ed. 2020. Accessed September 3, 2021. msif.org/resource/atlas-of-ms-2020.
2. Karampampa K, Gustavsson A, Miltenburger C, Tyas D. Treatment experience, burden and unmet needs (TRIBUNE) in MS study: results from the United Kingdom. Mult Scler. 2012;18(2_suppl):41-45
3. Thompson A, Kobelt G, Berg J, et al. New insights into the burden and costs of multiple sclerosis in Europe: Results for the United Kingdom. Mult Scler. 2017;23 (2_suppl):204-216.
4. Chataway J, Murphy N, Khurana V, Schofield H, Findlay J, Adlard N. Secondary progressive multiple sclerosis: a systematic review of costs and health state utilities. Curr Med Res Opin. 2021;37(6):995-1004.
5. Ontaneda D, Fox RJ, Chataway J. Clinical trials in progressive multiple sclerosis: lessons learned and future perspectives. Lancet Neurol. 2015;14(2):208-223.
6. Pardini M, Cutter G, Sormani MP. Clinical trial design for progressive MS trials. Mult Scler. 2017;23(12):1642-1648.
7. Sydes MR, Parmar MK, Mason MD, et al. Flexible trial design in practice: stopping arms for lack-of-benefit and adding research arms mid-trial in STAMPEDE: a multi-arm multi-stage randomized controlled trial. Trials 2012;13(1):168.
8. Phillips PPJ, Gillespie SH, Boeree M, et al. Innovative trial designs are practical solutions for improving the treatment of tuberculosis. J Infect Dis. 2012;205 (suppl_2): S250-S257.
9. Aisen PS, Bateman RJ, Carrillo M, et al. Platform trials to expedite drug development in Alzheimer’s disease: a report from the EU/US CTAD task force. J Prev Alzheimers Dis. 2021;8(3):306-312.
10. Zeissler ML, Li V, Parmar MKB, Carroll CB. Is it possible to conduct a multi-arm multi-stage platform trial in Parkinson’s disease: lessons learned from other neurodegenerative disorders and cancer. JPD 2020;10(2):413-428.
11. The University of Edinburgh. Motor neurone disease: a systematic multi-arm adaptive randomised trial. 2021. Accessed September 4, 2021. clinicaltrials.gov/ct2/ show/NCT04302870.
12. Kiernan MC, Vucic S, Talbot K, et al. Improving clinical trial outcomes in amyotrophic lateral sclerosis. Nat Rev Neurol. 2021;17 (2):104-118.
13. Parmar MK, Sydes MR, Cafferty FH, et al. Testing many treatments within a single protocol over 10 years at MRC Clinical Trials Unit at UCL: multi-arm, multi-stage platform, umbrella and basket protocols. Clin Trials. 2017;14(5):451-461.
14. Cunniffe N, Vuong KA, Ainslie D, et al. Systematic approach to selecting licensed drugs for repurposing in the treatment of progressive multiple sclerosis. J Neurol Neurosurg Psychiatry. 2021;92(3):295-302.
15. Sydes MR, Parmar MKB, James ND, et al. Issues in applying the multi-arm multi-stage methodology to a clinical trial in prostate cancer: the MRC STAMPEDE trial. Trials. 2009;10:39.
16. Chataway J, Schuerer N, Alsanousi A, et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet. 2014;383(9936):2213-2221.
17. Kapoor R, Ho PR, Campbell N, et al. Effect of natalizumab on disease progression in secondary progressive multiple sclerosis (ASCEND): a phase 3, randomised, double-blind, placebo-controlled trial with an open-label extension. Lancet Neurol. 2018;17(5):405-415.
18. Wason J, Magirr D, Law M, Jaki T. Some recommendations for multi-arm multi-stage trials. Stat Methods Med Res. 2016;25(2):716-727.
19. Tur C, Moccia M, Barkhof F, et al. Assessing treatment outcomes in multiple sclerosis trials and in the clinical setting. Nat Rev Neurol. 2018;14(2):75-93.
20. Meyer-Moock S, Feng YS, Maeurer M, Dippel FW, Kohlmann T. Systematic literature review and validity evaluation of the Expanded Disability Status Scale (EDSS) and the Multiple Sclerosis Functional Composite (MSFC) in patients with multiple sclerosis. BMC Neurol. 2014;14(1):58.
21. Cadavid D, Cohen JA, Freedman MS, et al. The EDSS-Plus, an improved endpoint for disability progression in secondary progressive multiple sclerosis. Mult Scler. 2017;23(1):94-105.
22. Lublin F, Miller DH, Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10023): 1075-1084.
23. The Queen Mary University of London. Chariot MS: a national (UK) phase IIb, multi-centre, randomised, double-blind, placebo-controlled (1:1) efficacy trial with cost-utility analysis of cladribine tablets (3.5 mg/kg over two years) in people with advanced multiple sclerosis: is cladribine superior to placebo in protecting upper limb function? 2021. Accessed September 30, 2021. clinicaltrials.gov/ct2/show/ NCT04695080.
24. Chataway J, De Angelis F, Connick P, et al. Efficacy of three neuroprotective drugs in secondary progressive multiple sclerosis (MS-SMART): a phase 2b, multiarm, double-blind, randomised placebo-controlled trial. Lancet Neurol. 2020;19(3):214-225.
25. Fox RJ, Coffey CS, Conwit R, et al. Phase 2 trial of ibudilast in progressive multiple sclerosis. N Engl J Med. 2018;379(9):846-855.
26. Sormani MP, Arnold DL, De Stefano N. Treatment effect on brain atrophy correlates with treatment effect on disability in multiple sclerosis. Ann Neurol. 2014;75 (1):43-49.
27. Moccia M, de Stefano N, Barkhof F. Imaging outcome measures for progressive multiple sclerosis trials. Mult Scler. 2017;23(12): 1614-1626.
28. Moccia M, Prados F, Filippi M, et al. Longitudinal spinal cord atrophy in multiple sclerosis using the generalized boundary shift integral. Ann Neurol. 2019;86(5):704-713.
29. Barro C, Benkert P, Disanto G, et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain. 2018;141(8):2382-2391.
30. Williams T, Zetterberg H, Chataway J. Neurofilaments in progressive multiple sclerosis: a systematic review. J Neurol. 2021;268(9):3212-3222.
31. Williams TE, BChir M, Holdsworth KP, et al. Assessing neurofilaments as biomarkers of neuroprotection in progressive multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2021;9:e1130.
32. Wason JMS, Brocklehurst P, Yap C. When to keep it simple: adaptive designs are not always useful. BMC Med. 2019;17(1):152.
33. Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, phase 3 study. Lancet. 2018;391(10127):1263-1273.
34. Freidlin B, Korn EL, Gray R, Martin A. Multi-arm clinical trials of new agents: some design considerations. Clin Cancer Res. 2008;14(14):4368-4371.
35. Dunnett CW. A multiple comparison procedure for comparing several treatments with a control. J Am Stat Assoc. 1955;50 (272):1096-1121.
36. University College London. A phase 3 randomised, double-blind, a clinical trial investigating the effectiveness of repurposed simvastatin compared to placebo in secondary progressive multiple sclerosis in slowing the progression of disability. 2018. Accessed July 1, 2021. clinicaltrials.gov/ct2/show/NCT03387670.
37. Chataway J, Nicholas R, Todd S, et al. A novel adaptive design strategy increases the efficiency of clinical trials in secondary progressive multiple sclerosis. Mult Scler. 2011;17(1):81-88.
38. Kahan BC, Forbes AB, Doré CJ, Morris TP. A re-randomisation design for clinical trials. BMC Med Res Methodol. 2015;15(1):96.
39. Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K. Alzheimer’s disease drug development pipeline. Alzheimer Dement Transl Res Clin Interv. 2020;6(1):e12050.
40. Mitsumoto H, Brooks BR, Silani V. Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trial be improved?. Lancet Neurol. 2014;13 (11):1127-1138.
41. Salloway S, Farlow M, McDade E, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat Med. 2021;27(7):1187-1196.
42. Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med. 2016;376(3):209-220.
43. Brochet B, Deloire MSA, Perez P, et al. Double-Blind controlled randomized trial of cyclophosphamide versus methylprednisolone in secondary progressive multiple sclerosis. PLoS One. 2017;12(1):e0168834.
44. Zajicek J, Ball S, Wright D, et al. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol. 2013;12(9):857-865.
45. Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460-471.
46. Kapoor R, Furby J, Hayton T, et al. Lamotrigine for neuroprotection in secondary progressive multiple sclerosis: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Neurol. 2010;9(7):681-688.
47. Spain R, Powers K, Murchison C, et al. Lipoic acid in secondary progressive MS. Neurol Neuroimmunol Neuroinflamm. 2017; 4(5):e374.
48. Tourbah A, Lebrun-Frenay C, Edan G, et al. MD1003 (high-dose biotin) for the treatment of progressive multiple sclerosis: a randomised, double-blind, placebo-controlled study. Mult Scler. 2016;22(13):1719-1731.
49. Kosa P, Wu T, Phillips J, et al. Idebenone does not inhibit disability progression in primary progressive MS. Mult Scler Relat Disord. 2020;45:102434.
50. Giovannoni G, Knappertz V, Steinerman JR, et al. A randomized, placebo-controlled, phase 2 trial of laquinimod in primary progressive multiple sclerosis. Neurology 2020;95(8):e1027-e1040.
CREDITS: Vivien Li, Baptiste Leurent, Frederik Barkhof, Marie Braisher, Fay Cafferty, Olga Ciccarelli, Arman Eshaghi, Emma Gray, Jennifer M. Nicholas, Mahesh Parmar, Guy Peryer, Jenny Robertson, Nigel Stallard, James Wason, Jeremy Chataway Neurology May 2022, 98 (18) 754-764; DOI: 10.1212/WNL.0000000000200604






