

A. M. Brandow & R. I. Liem
Abstract
Sickle cell disease (SCD), which affects approximately 100,000 individuals in the USA and more than 3 million worldwide, is caused by mutations in the βb globin gene that result in sickle hemoglobin production. Sickle hemoglobin polymerization leads to red blood cell sickling, chronic hemolysis and vaso-occlusion. Acute and chronic pain, as well as end-organ damage, occur throughout the lifespan of individuals living with SCD resulting in significant disease morbidity and a median life expectancy of 43 years in the USA. In this review, we discuss advances in the diagnosis and management of four major complications: acute and chronic pain, cardiopulmonary disease, central nervous system disease and kidney disease. We also discuss advances in disease-modifying and curative therapeutic options for SCD. The recent availability of l-glutamine, crizanlizumab and voxelotor provide an alternative or supplement to hydroxyurea, which remains the mainstay for disease-modifying therapy. Five-year event-free and overall survival rates remain high for individuals with SCD undergoing allogeneic hematopoietic stem cell transplant using matched sibling donors. However, newer approaches to graft-versus-host (GVHD) prophylaxis and the incorporation of post-transplant cyclophosphamide have improved engraftment rates, reduced GVHD and have allowed for alternative donors for individuals without an HLA-matched sibling. Despite progress in the field, additional longitudinal studies, clinical trials as well as dissemination and implementation studies are needed to optimize outcomes in SCD.
Introduction
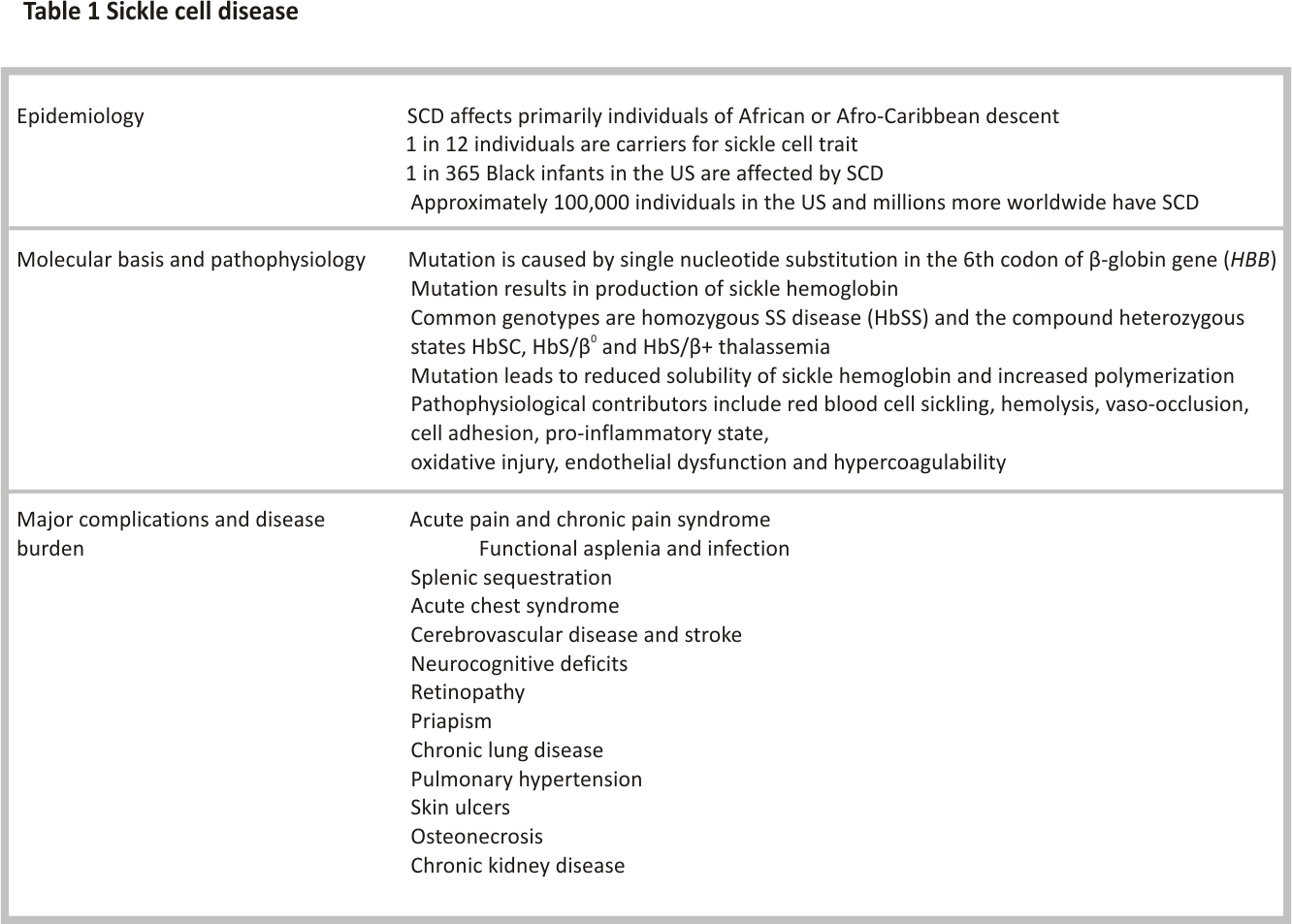
Sickle cell disease (SCD), a group of inherited hemoglobinopathies characterized by mutations that affect the β-globin chain of hemoglobin, affects approximately 100,000 people in the USA and more than 3 million people worldwide 1,2. SCD is characterized by chronic hemolytic anaemia, severe acute and chronic pain as well as end-organ damage that occurs across the lifespan. SCD is associated with premature mortality with a median age of death of 43 years (IQR 31.5–55 years)3. Treatment requires early diagnosis, prevention of complications and management of end-organ damage. In this review, we discuss recent advances in the diagnosis and management of four major complications in SCD: acute and chronic pain, cardiopulmonary disease, central nervous system disease and kidney disease. Updates in disease-modifying and curative therapies for SCD are also discussed.
Molecular basis and pathophysiology
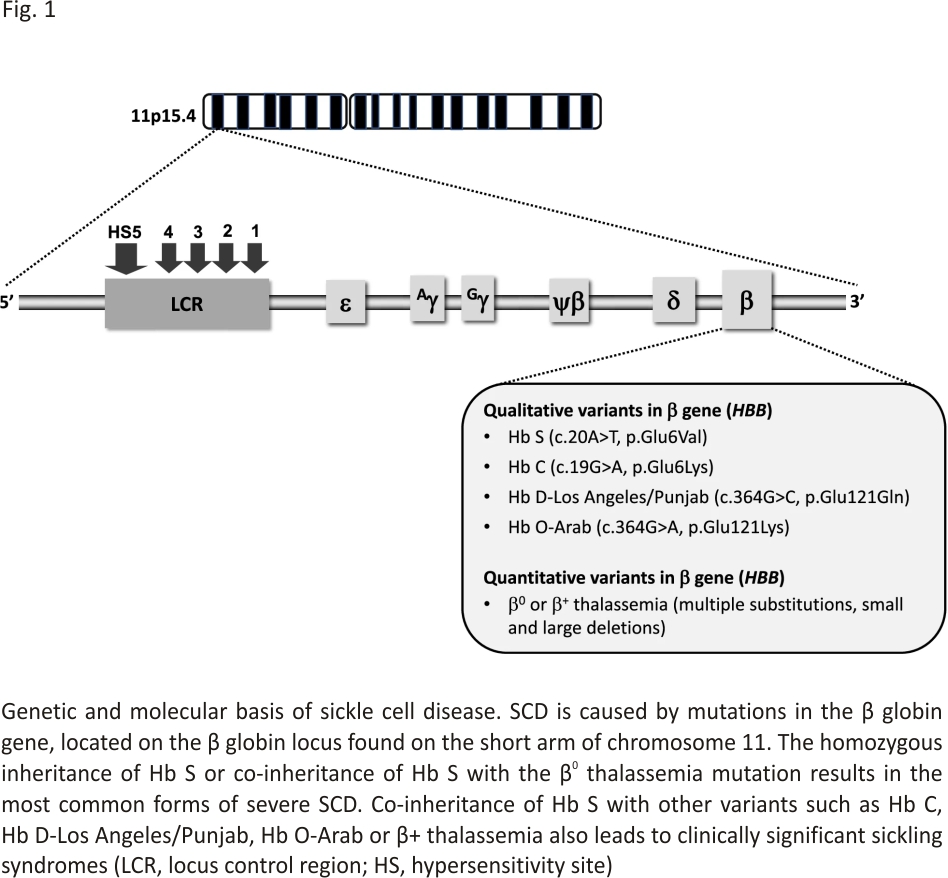
Hemoglobin S (HbS) results from the replacement of glutamic acid with valine in the sixth position of the β-globin chain of hemoglobin (Fig. 1). Severe forms of SCD include hemoglobin SS due to homozygous inheritance of HbS and S/β0 thalassemia due to co-inheritance of HbS with the β0 thalassemia mutation. Other forms include co-inheritance of HbS with other β-globin gene mutations such as hemoglobin C, hemoglobin D-Los Angeles/Punjab or β+ thalassemia. Hb S has reduced solubility and increased polymerization, which causes red blood cell sickling, hemolysis and vaso-occlusion (Table 1) that subsequently lead to pain episodes and end-organ damage such as cardiopulmonary, cerebrovascular and kidney disease (Table 2).
Acute and chronic pain
Severe intermittent acute pain is the most common SCD complication and accounts for over 70% of acute care visits for individuals with SCD 4. Chronic daily pain increases with older age, occurring in 30–40% of adolescents and adults with SCD 5, 6. Acute pain is largely related to vaso-occlusion of sickled red blood cells with ischemia-reperfusion injury and tissue infarction and presents in one isolated anatomic location (e.g., arm, leg, back) or multiple locations. Chronic pain can be caused by sensitization of the central and/or peripheral nervous system and is often diffuse with neuropathic pain features 7,8. A consensus definition for chronic pain includes “Reports of ongoing pain on most days over the past 6 months either in a single location or multiple locations”9. Disease complications such as avascular necrosis (hip, shoulder) and leg ulcers also cause chronic pain 9.
Diagnosis of acute and chronic pain
The gold standard for pain assessment and diagnosis is patient self-report. There are no reliable diagnostic tests to confirm the presence of acute or chronic pain in individuals with SCD except when there are identifiable causes like avascular necrosis on imaging or leg ulcers on the exam. The effects of pain on individuals’ function are assessed using patient-reported outcome measures (PROs) that determine to what extent pain interferes with individuals’ daily function. Tools shown to be valid, reliable and responsive can be used in clinical practice to track patients’ pain-related function overtime to determine additional treatment needs and to compare to population norms10. There are currently no plasma pain biomarkers that improve the assessment and management of SCD acute or chronic pain.
Depression and anxiety as co-morbid conditions in SCD can contribute to increased pain, more pain-related distress/interference and poor coping 11. The prevalence of depression and anxiety range from 26–33% and 6.5–36%, respectively, in adults with SCD 11,12,13. Adults with SCD have an 11% higher prevalence of depression compared to Black American adults without SCD14. Depression and anxiety can be assessed using self-reported validated screening tools (e.g., Depression: Patient Health Questionnaire (PHQ-9)15 for adults, Center for Epidemiologic Studies Depression Scale for Children (CES-DC) 16, PROMIS assessments for adults and children; Anxiety: Generalized Anxiety Disorder 7-item (GAD-7) scale for adults, State-Trait Anxiety Inventory for Children (STAIC) 17, PROMIS assessments for adults and children). Individuals who screen positive using these tools should be referred for evaluation by a psychologist/psychiatrist.
Management of acute and chronic pain
The goal of acute pain management is to provide sufficient analgesia to return patients to their usual function, which may mean complete resolution of pain for some or return to baseline chronic pain for others. The goal of chronic pain management is to optimize individuals’ function, which may not mean being pain-free. When there is an identifiable cause of chronic pain, treatment of the underlying issue (e.g., the joint replacement for avascular necrosis, leg ulcer treatment) is important. Opioids, oral for outpatient management and intravenous for inpatient management, are first-line therapy for acute SCD pain. In the acute care setting, analgesics should be initiated within 30–60 min of triage 18. Ketamine, a non-opioid analgesic, can be prescribed at sub-anaesthetic (analgesic) intravenous doses (0.1–0.3 mg/kg per h, maximum 1 mg/kg per h) as adjuvant treatment for acute SCD pain refractory to opioids 18,19. In an uncontrolled observational study of 85 patients with SCD receiving ketamine infusions for acute pain, ketamine was associated with a decrease in mean opioid consumption by oral morphine equivalents (3.1 vs. 2.2 mg/kg/day, p < 0.001) and reductions in mean pain scores (0–10 scale) from baseline until discontinuation of the infusion (7.81 vs. 5.44, p < 0.001) 20. Nonsteroidal anti-inflammatory drugs (NSAIDs) are routinely used as adjuvant therapy for acute pain treatment 18. In a RCT (n = 20) of hospitalized patients with acute pain, ketorolac was associated with lower total dose of meperidine required (1866.7 ± 12.4 vs. 2804.5 ± 795.1 mg, p < 0.05) and shorter hospitalization (median 3.3 vs. 7.2 days, p = 0.027) 21. In a case series of children treated for 70 acute pain events in the ED, 53% of events resolved with ketorolac and hydration alone with the reduction in 100 mm visual analogue scale (VAS) pain score from 60 to 13 (p < 0.001) 22. Patients at risk for NSAID toxicity (e.g., renal impairment, anticoagulation) should be identified.
Despite the paucity of data, chronic opioid therapy (COT) can be considered after assessing benefits versus harms 23 and the functional status of patients with SCD who have chronic pain. Harms of COT seen in patient populations other than SCD are dose-dependent and include myocardial infarction, bone fracture, increased risk of motor vehicle collisions, sexual dysfunction and mortality 23. There are few published studies investigating non-opioid analgesics for chronic SCD pain 24,25,26. In a randomized trial of 39 participants, those who received Vitamin D experienced a range of 6–10 pain days over 24 weeks while those who received a placebo experienced 10–16 pain days, which was not significantly different 26. In phase 1, an uncontrolled trial of 18 participants taking trifluoperazine, an antipsychotic drug, 8 participants showed a 50% reduction in the VAS (10 cm horizontal line) pain score from baseline on at least 3 assessments over 24 h without severe sedation or supplemental opioid analgesics, 7 participants showed pain reduction on 1 assessment, and the remaining 3 participants showed no reduction 24. Although published data are not available for serotonin and norepinephrine reuptake inhibitors (SNRIs), gabapentinoids and tricyclic antidepressants (TCAs) in individuals with SCD, evidence supports their use in fibromyalgia, a chronic pain condition similar to SCD chronic pain in a mechanism. A Cochrane Review that included 10 RCTs (n = 6038) showed that the SNRIs milnacipran and duloxetine, compared to placebo, were associated with a reduction in pain 27. A systematic review and meta-analysis of 9 studies (n = 520) showed the TCA amitriptyline improved pain intensity and function 28. Finally, a meta-analysis of 5 RCTs (n = 1874) of the gabapentinoid pregabalin showed a reduction in pain intensity 29. Collectively, the indirect evidence from fibromyalgia supports the conditional recommendation in current SCD practice guidelines to consider these 3 drug classes for chronic SCD pain treatment 18. Standard formulary dosing recommendations should be followed and reported adverse effects considered.
Non-pharmacologic therapies (e.g., integrative, psychological-based therapies) are important components of SCD pain treatment. In a case-control study of 101 children with SCD and chronic pain referred for cognitive behavioural therapy (CBT) (57 CBT, 44 no CBT) 30, CBT was associated with a more rapid decrease in pain hospitalizations (estimate − 0.63, p < 0.05) and faster reduction in hospital days over time (estimate − 5.50, p < 0.05). Among 18 children who received CBT and completed PROs pre- and 12 months posttreatment, improvements were seen in mean pain intensity (5.47 vs. 3.76, p = 0.009; 0–10 numeric rating pain scale), functional disability (26.24 vs. 15.18, p < 0.001; 0–60 score range) and pain coping (8.00 vs. 9.65, p = 0.03; 3–15 score range) post-treatment 30. In 2 uncontrolled clinical trials, acupuncture was associated with a significant reduction in pain scores by 2.1 points (0–10 numeric pain scale) in 24 participants immediately after treatment 31 or a significant mean difference in pre-post pain scores of 0.9333 (0–10 numeric pain scale) (p < 0.000) after 33 acupuncture sessions 32.
Cardiopulmonary disease
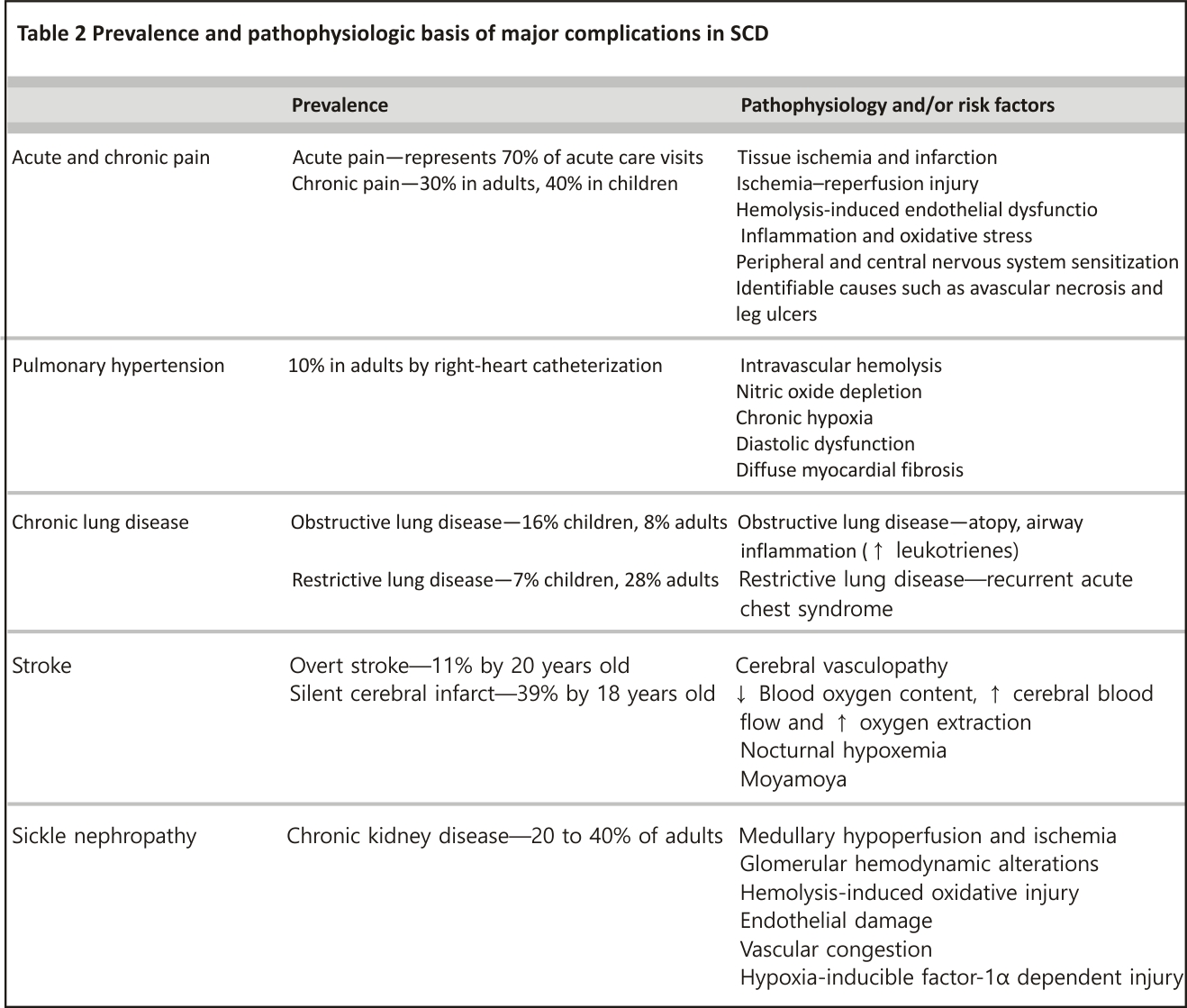
The cardiopulmonary disease is associated with increased morbidity and mortality in individuals with SCD. Pulmonary hypertension (PH), most commonly pulmonary arterial hypertension (PAH), is presently based on right-heart catheterization in up to 10% of adults with SCD 33. Chronic intravascular hemolysis represents the biggest risk factor for the development of PAH in SCD and leads to pulmonary arteriole vasoconstriction and smooth muscle proliferation. Based on pulmonary function testing (PFT), obstructive lung disease may be observed in 16% of children and 8% of adults with SCD, while restrictive lung disease may be seen in up to 28% of adults and only 7% of children with SCD 34,35. Sleep-disordered breathing, which can manifest as obstructive sleep apnea or nocturnal hypoxemia, occurs in up to 42% of children and 46% of adults with SCD 36,37. Cardiopulmonary disease, including PH or restrictive lung disease, presents with dyspnea with or without exertion, chest pain, hypoxemia or exercise intolerance that is unexplained or increased from baseline. Obstructive lung disease can also present with wheezing.
Diagnosis of cardiopulmonary disease
The confirmation of PH in patients with SCD requires right-heart catheterization. Recently, the mean pulmonary artery pressure threshold used to define PH in the general population was lowered from ≥25 to ≥20 mm Hg 38. Elevated peak tricuspid regurgitant jet velocity (TRJV)≥2.5 m/s on Doppler echocardiogram (ECHO) is associated with early mortality in adults with SCD and may suggest elevated pulmonary artery pressures, especially when other signs of PH (e.g., right-heart strain, septal flattening) or left ventricular diastolic dysfunction, which may contribute to PH, are present 39. However, the positive predictive value (PPV) of peak TRJV alone for identifying PH in adults with SCD is only 25% 40. Increasing the peak TRJV threshold to at least 2.9 m/s has been shown to increase the PPV to 64%. For a peak TRJV of 2.5–2.8 m/s, an increased N-terminal pro-brain natriuretic peptide (NT-proBNP) ≥164.5 pg/mL or a reduced 6-min walk distance (6MWD) < 333 m can also improve the PPV to 62% with a false-negative rate of 7% 33, 40, 41.
PFT, which includes spirometry and measurement of lung volumes and diffusion capacity, is standard for diagnosing obstructive and restrictive lung disease in patients with SCD. Emerging modalities include impulse oscillometry, a non-invasive method using forced sound waves to detect changes in lower airway mechanics in individuals unable to perform spirometry 42, and airway provocation studies using cold air or methacholine to reveal latent airway hyperreactivity 43. Formal in-lab, sleep study/polysomnography remains the gold standard to evaluate for sleep-disordered breathing, which may include nocturnal hypoxemia, apnea/hypopnea events and other causes of sleep disruption. Nocturnal hypoxemia may increase red blood cell sickling, cellular adhesion and endothelial dysfunction. In 47 children with SCD, mean overnight oxygen saturation was higher in those with grade 0 compared to grade 2 or 3 cerebral arteriopathy (97 ± 1.6 vs. 93.9 ± 3.7 vs. 93.5 ± 3.0%, p < 0.01) on magnetic resonance angiography and lower overnight oxygen saturation was independently associated with mild, moderate or severe cerebral arteriopathy after adjusting for reticulocytosis (OR 0.50, 95% CI 0.26– 0.96, p < 0.05) 44.
Management of cardiopulmonary disease
Patients with SCD who have symptoms suggestive of cardiopulmonary diseases, such as worsening dyspnea, hypoxemia or reduced exercise tolerance, should be evaluated with a diagnostic ECHO and PFT. The presence of snoring witnessed apnea, respiratory pauses or hypoxemia during sleep, daytime somnolence or nocturnal enuresis in older children and adults is sufficient for a diagnostic sleep study.
Without treatment, the mortality rate in SCD patients with PH is high compared to those without (5-year, an all-cause mortality rate of 32 vs. 16%, p < 0.001)33. PAH-targeted therapies should be considered for SCD patients with PAH confirmed by right-heart catheterization. However, the only RCT (n = 6) in individuals with SCD and PAH confirmed by right-heart catheterization (bosentan versus placebo) was stopped early for poor accrual with no efficacy endpoints analyzed 45. In SCD patients with elevated peak TRJV, a randomized controlled trial (n = 74) of sildenafil, a phosphodiesterase-5 inhibitor, was discontinued early due to increased pain events in the sildenafil versus placebo arm (35 vs. 14%, p = 0.029) with no treatment benefit 46. Despite the absence of clinical trial data, patients with SCD and confirmed PH should be considered for hydroxyurea or monthly red blood cell transfusions are given their disease-modifying benefits. In a retrospective analysis of 13 adults with SCD and PAH, 77% of patients starting at a New York Heart Association (NYHA) functional capacity class III or IV achieved class I/II after a median of 4 exchange transfusions with improvement in median pulmonary vascular resistance (3.7 vs. 2.8 Wood units, p = 0.01) 47.
Approximately 28% of children with SCD have asthma, which is associated with increased pain episodes that may result from impaired oxygenation leading to sickling and vaso-occlusion as well as with acute chest syndrome and higher mortality 48,49,50. First-line therapies include standard beta-adrenergic bronchodilators and supplemental oxygen as needed. When corticosteroids are indicated, courses should be tapered over several days given the risk of rebound SCD pain from abrupt discontinuation. Inhaled corticosteroids such as fluticasone propionate or beclomethasone disproportionate are reserved for patients with recurrent asthma exacerbations, but their anti-inflammatory effects and impact on preventing pain episodes in patients with SCD who do not have asthma are under investigation 51. Finally, the management of sleep-disordered breathing is tailored to the findings of a formal sleep study in consultation with a sleep/pulmonary specialist.
Central nervous system (CNS) complications
CNS complications, such as overt and silent cerebral infarcts, cause significant morbidity in individuals with SCD. Eleven per cent of patients with HbSS disease by age 20 years and 24% by age 45 years will have had an overt stroke 52. Silent cerebral infarcts occur in 39% by 18 years and in > 50% by 30 years 53,54. Patients with either type of stroke are at increased risk of recurrent stroke 55. Overt stroke involves large arteries, including middle cerebral arteries and intracranial internal carotid arteries, while silent cerebral infarcts involve penetrating arteries. The pathophysiology of overt stroke includes vasculopathy, increased sickled red blood cell adherence, hemolysis-induced endothelial activation and altered vasomotor tone 56. Overt strokes present as weakness or paresis, dysarthria or aphasia, seizures, sensory deficits, headache or altered level of consciousness, while silent cerebral infarcts are associated with cognitive deficits, including lower IQ and impaired academic performance.
Diagnosis of CNS complications in SCD
Overt stroke is diagnosed by evidence of acute infarct on brain MRI diffusion-weighted imaging and focal deficit on the neurologic exam. A silent cerebral infarct is defined by a brain “MRI signal abnormality at least 3 mm in one dimension and visible in 2 planes on fluid-attenuated inversion recovery (FLAIR) T2-weighted images” and no deficit on neurologic exam 57. Since silent cerebral infarcts cannot be detected clinically, a screening baseline brain MRI is recommended in school-aged children with SCD 58. Recent SCD clinical practice guidelines also suggest a screening brain MRI in adults with SCD to facilitate rehabilitation services, patient and family understanding of cognitive deficits and further needs assessment 58. An MRA should be added to screening /diagnostic MRIs to evaluate for cerebral vasculopathy (e.g., moyamoya), which may increase the risk for recurrent stroke or hemorrhage 59.
Annual screening for increased stroke risk by transcranial doppler (TCD) ultrasound is recommended by the American Society of Hematology for children 2–16 years old with HbSS or HbS/β° thalassemia 58. Increased stroke risk on non-imaging TCD is indicated by abnormally elevated cerebral blood flow velocity, defined as ≥ 200 cm/s (time-averaged mean of the maximum velocity) on 2 occasions or a single velocity of > 220 cm/s in the distal internal carotid or proximal middle cerebral artery 60. Many centres rely on imaging TCD, which results in velocities 10–15% lower than values obtained by non-imaging protocols and therefore, require adjustments to cut-offs for abnormal velocities. Data supporting stroke risk assessment using TCD are lacking for adults with SCD and standard recommendations do not exist.
Neurocognitive deficits occur in over 30% of children and adults with severe SCD 61,62. These occur as a result of overt and/or silent cerebral infarcts but in some patients, the aetiology is unknown. The Bright Futures Guidelines for Health Supervision of Infants, Children and Adolescents or the Cognitive Assessment Toolkit for adults are commonly used tools to screen for developmental delays or neurocognitive impairment 58. Abnormal results should prompt referral for formal neuropsychological evaluation, which directs the need for brain imaging to evaluate for silent cerebral infarcts and facilitate educational/vocational accommodations.
Management of CNS complications
Monthly chronic red blood cell transfusions to suppress HbS < 30% are standard of care for primary stroke prevention in children with an abnormal TCD. In an RCT of 130 children, chronic transfusions, compared to no transfusions, were associated with a difference in stroke risk of 92% (1 vs. 10 strokes, p < 0.001) 60. However, children with abnormal TCD and no MRI/MRA evidence of cerebral vasculopathy can safely transition to hydroxyurea after 1 year of transfusions 63. Lifelong transfusions to maintain HbS < 30% remain the standard of care for secondary stroke prevention in individuals with overt stroke 64. Chronic monthly red blood cell transfusions should also be considered for children with silent cerebral infarct 58. In a randomized controlled trial (n = 196), monthly transfusions, compared to observation without hydroxyurea, reduced the risk of overt stroke, new silent cerebral infarct or enlarging silent cerebral infarct in children with HbSS or HbS/β0 thalassemia and an existing silent cerebral infarct (2 vs. 4.8 events, incidence rate ratio of 0.41, 95% CI 0.12–0.99, p = 0.04) 57.
Acute stroke treatment requires transfusion therapy to increase cerebral oxygen delivery. Red blood cell exchange transfusion, defined as the replacement of patients’ red blood cells with donor red blood cells, to rapidly reduce HbS to < 30% is the recommended treatment as simple transfusion alone is shown to have a fivefold greater relative risk (57 vs. 21% with recurrent stroke, RR = 5.0; 95% CI 1.3–18.6) of subsequent stroke compared to exchange transfusion 65. However, a simple transfusion is often given urgently while preparing for an exchange transfusion 58. Tissue plasminogen activator (tPA) is not recommended for children with SCD who have an acute stroke since the pathophysiology of SCD stroke is less likely to be thromboembolic in origin and there is a risk for harm. Since the benefits and risks of tPA in adults with SCD and overt stroke are not clear, its use depends on comorbidities, risk factors and stroke protocols but should not delay or replace prompt transfusion therapy.
Data guiding treatment of SCD cerebral vasculopathy (e.g., moyamoya) are limited, and only nonrandomized, low-quality evidence exists for neurosurgical interventions (e.g., encephaloduroarteriosynangiosis) 66. Consultation with a neurosurgeon to discuss surgical options in patients with moyamoya and a history of stroke or transient ischemic attack should be considered 58.
Kidney disease
Glomerulopathy, characterized by hyperfiltration leading to albuminuria, is an early asymptomatic manifestation of SCD nephropathy and worsens with age. Hyperfiltration, defined by an absolute increase in glomerular filtration rate, may be seen in 43% of children with SCD 67. Albuminuria, defined by the presence of urine albumin ≥ 30 mg/g over 24 h, has been observed in 32% of adults with SCD 68. Glomerulopathy results from intravascular hemolysis and endothelial dysfunction in the renal cortex. Medullary hypoperfusion and ischemia also contribute to kidney disease in SCD, causing hematuria, urine concentrating defects and distal tubular dysfunction 69. Approximately 20–40% of adults with SCD develop chronic kidney disease (CKD) and are at risk of developing the end-stage renal disease (ESRD), with rapid declines in estimated glomerular filtration rate (eGFR) > 3 mL/min/1.73 m2 associated with increased mortality (HR 2.4, 95% CI 1.31–4.42, p = 0.005) 68.
Diagnosis of kidney disease in SCD
The diagnosis of sickle cell nephropathy is made by detecting abnormalities such as albuminuria, hematuria or CKD rather than by distinct diagnostic criteria in SCD, which have not been developed. Traditional markers of kidney function such as serum creatinine and eGFR should be interpreted with caution in individuals with SCD because renal hyperfiltration affects their accuracy by increasing both. Practical considerations preclude directly measuring GFR by urine or plasma clearance techniques, which achieves the most accurate results. The accuracy of eGFR, however, may be improved by equations that incorporate serum cystatin C 70.
Since microalbuminuria/proteinuria precedes CKD in SCD, annual screening for urine microalbumin/protein is recommended beginning at the age of 10 years 71. When evaluating urine for microalbumin concentration, samples from the first morning rather than random voids are preferable to exclude orthostatic proteinuria. Recent studies suggest HMOX1 and APOL1 gene variants may be associated with CKD in individuals with SCD 72. Potential novel predictors of acute kidney injury in individuals with SCD include urine biomarkers kidney injury molecule 1 (KIM-1) 73, monocyte chemotactic protein 1 (MCP-1) 74 and neutrophil gelatinase-associated lipocalin (NGAL) 75. Their contribution to chronic kidney disease and interaction with other causes of kidney injury in SCD (e.g., inflammation, hemolysis) is not clear.
Management of kidney disease
Managing kidney complications in SCD should focus on mitigating risk factors for acute and chronic kidney injury such as medication toxicity, reduced kidney perfusion from hypotension and dehydration, and general disease progression, as well as early screening and treatment of microalbuminuria/proteinuria. Acute kidney injury, either an increase in serum creatinine ≥ 0.3 mg/dL or a 50% increase in serum creatinine from baseline, is associated with ketorolac use in children with SCD hospitalized for pain 76. Increasing intravenous fluids to maintain urine output > 0.5 to 1 mL/kg/h and limiting NSAIDs and antibiotics associated with nephrotoxicity in this setting are important. Despite the absence of controlled clinical trials, hydroxyurea may be associated with improvements in glomerular hyperfiltration and urine concentrating ability in children with SCD 77,78. Hydroxyurea is also associated with a lower prevalence (34.7 vs. 55.4%, p = 0.01) and the likelihood of albuminuria (OR 0.28, 95% CI 0.11–0.75, p = 0.01) in adults with SCD after adjusting for age, angiotensin-converting enzyme inhibitor (ACE-I)/angiotensin receptor blockade (ARB) use and major disease risk factors 79.
ACE-I or ARB therapy reduces microalbuminuria in patients with SCD. In a phase 2 trial of 36 children and adults, a ≥ 25% reduction in urine albumin-to-creatinine ratio was observed in 83% (p < 0.0001) and 58% (p < 0.0001) of patients with macroalbuminuria (> 300 mg/g creatinine) and microalbuminuria (30–300 mg/g creatinine), respectively, after 6 months of treatment with losartan at a dose of 0.7 mg/kg/day (max of 50 mg) in children and 50 mg daily in adults 80. However, ACE-I or ARB therapy has not been shown to improve kidney function or prevent CKD. Hemodialysis is associated with a 1-year mortality rate of 26.3% after starting hemodialysis and an increased risk of death in SCD patients with ESRD compared to non-SCD patients with ESRD (44.6 vs. 34.5% deaths, the mortality hazard ratio of 2.8, 95% CI 2.31–3.38) 81. Renal transplant should be considered for individuals with SCD and ESRD because of recent improvements in renal graft survival and post-transplant mortality 82.
Disease-modifying therapies in SCD
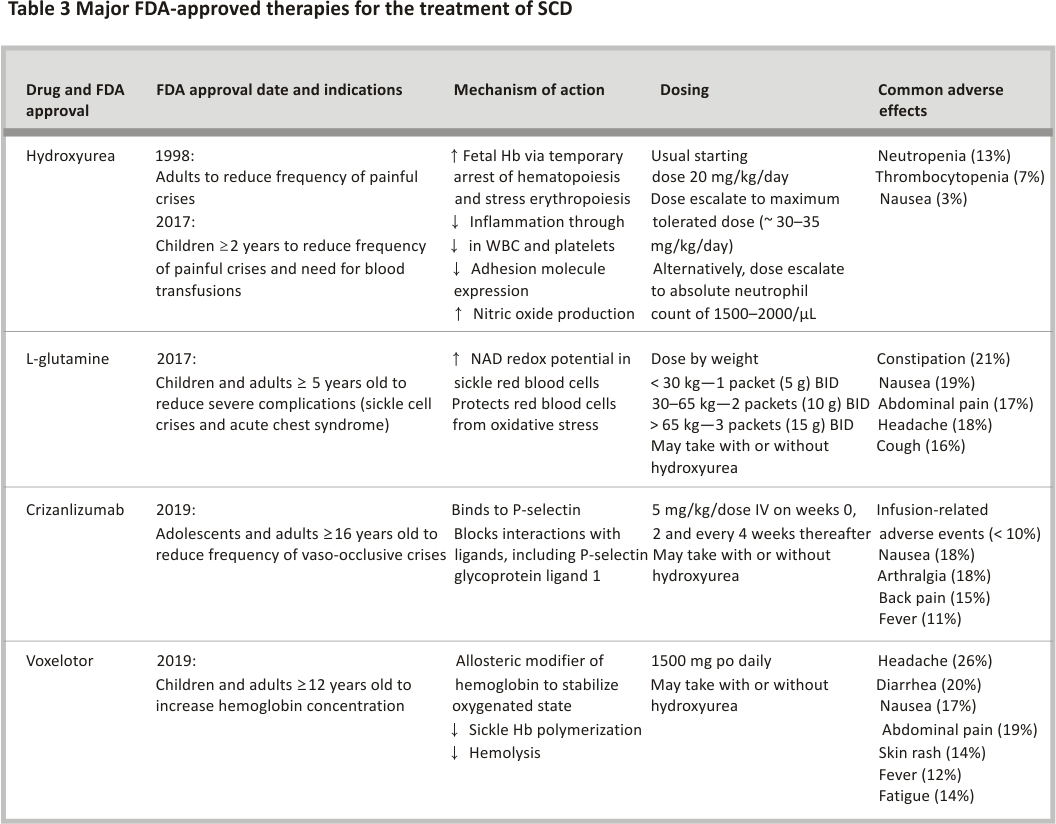
Since the publication of its landmark trial in 1995, hydroxyurea continues to represent a mainstay of disease-modifying therapy for SCD. Hydroxyurea induces fetal hemoglobin production through stress erythropoiesis, reduces inflammation, increases nitric oxide and decreases cell adhesion. The FDA approved hydroxyurea in 1998 for adults with SCD. Subsequently, hydroxyurea was FDA approved for children in 2017 to reduce the frequency of pain events and the need for blood transfusions in children ≥ 2 years of age 63. The landscape of disease-modifying therapies, however, has improved with the recent FDA approval of 3 other treatments—l-glutamine and crizanlizumab for reducing acute complications (e.g., pain), and voxelotor for improving anaemia (Table 3) 83,84,85. Other therapies in current development focus on inducing fetal hemoglobin, reducing anti-sickling or cellular adhesion, or activating pyruvate kinase-R.
L-glutamine
Glutamine is required for the synthesis of glutathione, nicotinamide adenine dinucleotide and arginine. The essential amino acid protects red blood cells against oxidative damage, which forms the basis for its proposed utility in SCD. The exact mechanism of benefit in SCD, however, remains unclear. In a phase 3 RCT of 230 participants (hemoglobin SS or S/β0 thalassemia), l-glutamine compared to placebo was associated with fewer pain events (median 3 vs. 4, p = 0.005) and hospitalizations for pain (median 2 vs. 3, p = 0.005) over the 48-week treatment period 84. The percentage of patients who had at least 1 episode of acute chest syndrome, defined as the presence of chest wall pain with fever and a new pulmonary infiltrate, was lower in the l-glutamine group (8.6 vs. 23.1%, p = 0.003). There were no significant between-group differences in hemoglobin, hematocrit or reticulocyte count. Common side effects of l-glutamine include GI upset (constipation, nausea, vomiting and abdominal pain) and headaches.
Crizanlizumab
P-selectin expression, triggered by inflammation, promotes the adhesion of neutrophils, activated platelets and sickle red blood cells to the endothelial surface and to each other, which promotes vaso-occlusion in SCD. Crizanlizumab, given as a monthly intravenous infusion, is a humanized monoclonal antibody that binds P-selectin and blocks the adhesion molecule’s interaction with its ligand, P-selectin glycoprotein ligand 1. FDA approval for crizanlizumab was based on a phase 2 RCT (n = 198, all genotypes), in which the median rate of pain events (primary endpoint) was lower (1.63 vs. 2.68, p = 0.01) and time to first pain event (secondary endpoint) was longer (4.07 vs. 1.38 months, p = 0.001) for patients on high-dose crizanlizumab (5 mg/kg/dose) compared to placebo-treated for 52 weeks (14 doses total) 83. In this trial, patients with SCD on chronic transfusion therapy were excluded, but those on stable hydroxyurea dosing were not. Adverse events were uncommon but included headache, back pain, nausea, arthralgia and pain in the extremity.
Voxelotor
Polymerization of Hb S in the deoxygenated state represents the initial step in red blood cell sickling, which leads to reduced red blood cell deformability and increased hemolysis. Voxelotor is a first-in-class allosteric modifier of Hb S that increases oxygen affinity. The primary endpoint for the phase 3 RCT of voxelotor (n = 274, all genotypes) that led to FDA approval was an increase in hemoglobin of at least 1 g/dL after 24 weeks of treatment 85. More participants receiving 1500 mg daily of oral voxelotor versus placebo had a hemoglobin response of at least 1 g/dL (51%, 95% CI 41–61 vs. 7%, 95% CI 1–12, p < 0.001). Approximately 2/3 of the participants in these trials were on hydroxyurea, with treatment benefits observed regardless of hydroxyurea status. Despite improvements associated with voxelotor in biomarkers of hemolysis (reticulocyte count, indirect bilirubin and lactate dehydrogenase), the annualized incidence rate of the vaso-occlusive crisis was not significantly different among treatment groups. Adverse events included headaches, GI symptoms, arthralgia, fatigue and rash.
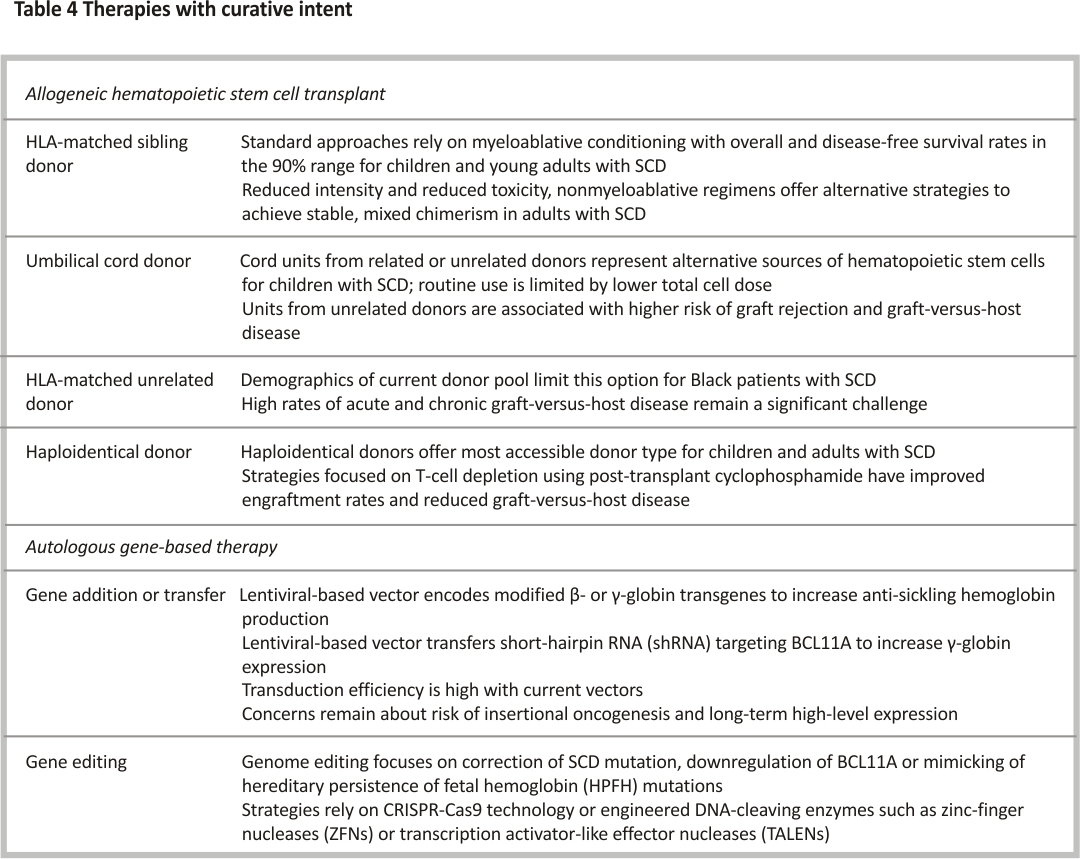
Curative therapies in SCD
For individuals with SCD undergoing hematopoietic stem cell transplantation (HSCT) using HLA-matched sibling donors and either myeloablative or reduced-intensity conditioning regimens, the five-year event-free and overall survival is high at 91% and 93%, respectively 86. The limited availability of HLA-matched sibling donors in this population requires alternative donors or the promise of autologous strategies such as gene-based therapies (i.e. gene addition, transfer or editing) (Table 4). Matched unrelated donors have not been used routinely due to the increased risk of graft-versus-host disease (GVHD) as high as 19% (95% CI 12–28) in the first 100 days for acute GVHD and 29% (95% CI 21–38) over 3 years for chronic GVHD 87. Haploidentical HSCT, using biological parents or siblings as donors, that incorporates post-transplant cyclophosphamide demonstrates acceptable engraftment rates, transplant-related morbidity and overall mortality 88. Regardless of allogeneic HSCT type, older age is associated with lower event-free (102/418 vs. 72/491 events, HR 1.74, 95% CI 1.24–2.45) and overall survival (54/418 vs. 22/491 events, HR 3.15, 95% CI 1.86–5.34) in patients ≥ 13 years old compared to < 12 years old undergoing HSCT 87.
Advancing research in SCD
Despite progress to date, additional high-quality, longitudinal data are needed to better understand the natural history of the disease and to inform optimal screening for SCD-related complications. In the era of multiple FDA-approved therapies with disease-modifying potential, clinical trials to evaluate additional indications and test them in combination with or compared to each other are needed. Dissemination and implementation studies are also needed to identify barriers and facilitators related to treatment in everyday life, which can be incorporated into decision aids and treatment algorithms for patients and their providers 89. Lastly, continued efforts should acknowledge social determinants of health and other factors that affect access and disease-related outcomes such as the role of third-party payers, provider and patient education, health literacy and patient trust. Establishing evidence-derived quality of care metrics can also drive public policy changes required to ensure care optimization for this population.
Conclusions
SCD is associated with complications that include acute and chronic pain as well as end-organ damage such as cardiopulmonary, cerebrovascular and kidney disease that results in increased morbidity and mortality. Several well-designed clinical trials have resulted in key advances in the management of SCD in the past decade. Data from these trials have led to FDA approval of 3 new drugs, l-glutamine, crizanlizumab and voxelotor, which prevent acute pain and improve chronic anaemia. Moderate to high-quality data support recommendations for managing SCD cerebrovascular disease and early kidney disease. However, further research is needed to determine the best treatment for chronic pain and cardiopulmonary disease in SCD. Comparative effectiveness research, dissemination and implementation studies and a continued focus on social determinants of health are also essential.
Availability of data and materials
Not applicable.
Abbreviations
6-MWD: Six-minute walk distance
ACE-I: Angiotensin-converting enzyme inhibitor
ARB: Angiotensin receptor blockade
CBT: Cognitive behavioural therapy
CKD: Chronic kidney disease
COT: Chronic opioid therapy
ECHO: Echocardiogram
ESRD: End-stage renal disease
FLAIR: Fluid-attenuated inversion recovery
GFR: Glomerular filtration rate
GVHD: Graft-versus-host disease
HbS: Hemoglobin S
HSCT: Hematopoietic stem cell transplant
NSAIDs: Nonsteroidal anti-inflammatory drugs
NT-proBNP: N-terminal pro-brain natriuretic peptide
NYHA: New York Heart Association
PAH: Pulmonary arterial hypertension
PFT: Pulmonary function test
PH: Pulmonary hypertension
PPV: Positive predictive value
PROs: Patient-reported outcomes
RCT: Randomized controlled trial
SCD: Sickle cell disease
SNRIs: Serotonin and norepinephrine reuptake inhibitors
TCAs: Tricyclic antidepressants
TCD: Transcranial Doppler
tPA: Tissue plasminogen activator
TRJV: Tricuspid regurgitant jet velocity
VAS: Visual Analog Scale
Acknowledgements
We would like to acknowledge Lana Mucalo, MD, for supporting data collection for this manuscript.
Funding
Not applicable.
Author information
Affiliations
Department of Pediatrics, Section of Pediatric Hematology/Oncology/Bone Marrow Transplantation, Medical College of Wisconsin, Milwaukee, WI, USA
A. M. Brandow
Division of Hematology, Oncology and Stem Cell Transplantation, Ann and Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL, USA
R. I. Liem
Contributions
AB and RL contributed equally to the writing and editing of this manuscript. All authors read and approved the final manuscript.
Corresponding author
Correspondence to R. I. Liem.
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no competing interests to declare.
References
1. Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013;381(9861):1 42–51.
2. Brousseau DC, Panepinto JA, Nimmer M, Hoffmann RG. The number of people with sickle-cell disease in the United States: national and state estimates. Am J Hematol. 2010;85(1):77–8.
3. Payne AB, Mehal JM, Chapman C, Haberling DL, Richardson LC, Bean CJ, et al. Trends in sickle cell disease-related mortality in the United States, 1979 to 2017. Ann Emerg Med. 2020;76(3S): S28–36.
4. Brousseau DC, Owens PL, Mosso AL, Panepinto JA, Steiner CA. Acute care utilization and rehospitalizations for sickle cell disease. JAMA. 2010;303(13):1 288–94.
5. Sil S, Cohen LL, Dampier C. Psychosocial and functional outcomes in youth with chronic sickle cell pain. Clin J Pain. 2016;32(6): 527–33.
6. Smith WR, Penberthy LT, Bovbjerg VE, McClish DK, Roberts JD, Dahman B, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med. 2008;148(2):94–101.
7. Ballas SK, Darbari DS. Neuropathy, neuropathic pain, and sickle cell disease. Am J Hematol. 2013;88(11):927–9.
8. Sharma D, Brandow AM. Neuropathic pain in individuals with sickle cell disease. Neurosci Lett. 2020;714:134445.
9. Dampier C, Palermo TM, Darbari DS, Hassell K, Smith W, Zempsky W. AAPT diagnostic criteria for chronic sickle cell disease pain. J Pain. 2017;18(5):490–8.
10. Darbari DS, Hampson JP, Ichesco E, Kadom N, Vezina G, Evangelou I, et al. Frequency of hospitalizations for pain and association with altered brain network connectivity in sickle cell disease. J Pain. 2015;16(11): 1077–86.
11. Levenson JL, Mcclish DK, Dahman BA, Bovbjerg VE, Penberthy LT, et al. Depression and anxiety in adults with sickle cell disease: the PiSCES project. Psychosom Med. 2008;70(2):192–6.
12. Treadwell MJBB, Kaur K, Gildengorin G. Emotional distress, barriers to care, and health-related quality of life in sickle cell disease. J Clin Outcomes Manag. 2015;22: 10.
13. Jonassaint CR, Jones VL, Leong S, Frierson GM. A systematic review of the association between depression and health care utilization in children and adults with sickle cell disease. Br J Haematol. 2016; 174(1):136–47.
14. Laurence B, George D, Woods D. Association between elevated depressive symptoms and clinical disease severity in African-American adults with sickle cell disease. J Natl Med Assoc. 2006;98(3):365–9.
15. Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9): 606–13.
16. Faulstich ME, Carey MP, Ruggiero L, Enyart P, Gresham F. Assessment of depression in childhood and adolescence: An evaluation of the Center for Epidemiological Studies Depression Scale for Children (CES-DC). Am J Psychiatry. 1986;143(8):1024–7.
17. Spielberger CD, Gorsuch RL, Lushene R, Vagg PR, Jacobs GA. Manual for the state-trait anxiety inventory. Palo Alto: Consulting Psychologists Press; 1983.
18. Brandow AM, Carroll CP, Creary S, Edwards-Elliott R, Glassberg J, Hurley RW, et al. American Society of Hematology 2020 guidelines for sickle cell disease: management of acute and chronic pain. Blood Adv. 2020;4(12):2656–701.
19. Lubega FA, DeSilva MS, Munube D, Nkwine R, Tumukunde J, Agaba PK, et al. Low dose ketamine versus morphine for acute severe vaso occlusive pain in children: a randomized controlled trial. Scand J Pain. 2018;18(1):19–27.
20. Nobrega R, Sheehy KA, Lippold C, Rice AL, Finkel JC, Quezado ZMN. Patient characteristics affect the response to ketamine and opioids during the treatment of vaso-occlusive episode-related pain in sickle cell disease. Pediatr Res. 2018;83(2):445–54.
21. Perlin E, Finke H, Castro O, Rana S, Pittman J, Burt R, et al. Enhancement of pain control with ketorolac tromethamine in patients with sickle cell vaso-occlusive crisis. Am J Hematol. 1994;46(1):43–7.
22. Beiter JL Jr, Simon HK, Chambliss CR, Adamkiewicz T, Sullivan K. Intravenous ketorolac in the emergency department management of sickle cell pain and predictors of its effectiveness. Arch Pediatr Adolesc Med. 2001;155(4):496 –500.
23. Chou R, Turner JA, Devine EB, Hansen RN, Sullivan SD, Blazina I, et al. The effectiveness and risks of long-term opioid therapy for chronic pain: a systematic review for a National Institutes of Health Pathways to Prevention Workshop. Ann Intern Med. 2015;162(4):276–86.
24. Molokie RE, Wilkie DJ, Wittert H, Suarez ML, Yao Y, Zhao Z, et al. Mechanism-driven phase I translational study of trifluoperazine in adults with sickle cell disease. Eur J Pharmacol. 2014;723:419–24.
25. Schlaeger JM, Molokie RE, Yao Y, Suarez ML, Golembiewski J, Wilkie DJ, et al. Management of sickle cell pain using pregabalin: a pilot study. Pain Manag Nurs. 2017;18(6):391–400.
26. Osunkwo I, Ziegler TR, Alvarez J, McCracken C, Cherry K, Osunkwo CE, et al. High dose vitamin D therapy for chronic pain in children and adolescents with sickle cell disease: results of a randomized double-blind pilot study. Br J Haematol. 2012;159 (2):211–5.
27. Hauser W, Urrutia G, Tort S, Uceyler N, Walitt B. Serotonin and noradrenaline reuptake inhibitors (SNRIs) for fibromyalgia syndrome. Cochrane Database Syst Rev. 2013;1:CD010292.
28. Hauser W, Wolfe F, Tolle T, Uceyler N, Sommer C. The role of antidepressants in the management of fibromyalgia syndrome: a systematic review and meta-analysis. CNS Drugs. 2012;26(4):297–307.
29. Derry S, Cording M, Wiffen PJ, Law S, Phillips T, Moore RA. Pregabalin for pain in fibromyalgia in adults. Cochrane Database Syst Rev. 2016;9:CD011790.
30. Sil S, Lai K, Lee JL, Gilleland Marchak J, Thompson B, Cohen L, et al. Preliminary evaluation of the clinical implementation of cognitive-behavioural therapy for chronic pain management in pediatric sickle cell disease. Complement Ther Med. 2020;49: 102348.
31. Lu K, Cheng MC, Ge X, Berger A, Xu D, Kato GJ, et al. A retrospective review of acupuncture use for the treatment of pain in sickle cell disease patients: a descriptive analysis from a single institution. Clin J Pain. 2014;30(9):825–30.
32. Mahmood LA, Reece-Stremtan S, Idiokitas R, Martin B, Margulies S, Hardy SJ, et al. Acupuncture for pain management in children with sickle cell disease. Complement Ther Med. 2020;49:102287.
33. Mehari A, Alam S, Tian X, Cuttica MJ, Barnett CF, Miles G, et al. Hemodynamic predictors of mortality in adults with sickle cell disease. Am J Respir Crit Care Med. 2013;187(8):840–7.
34. Cohen RT, Strunk RC, Rodeghier M, Rosen CL, Kirkham FJ, Kirkby J, et al. Pattern of lung function is not associated with prior or future morbidity in children with sickle cell anaemia. Ann Am Thorac Soc. 2016; 13(8):1314–23.
35. Kassim AA, Payne AB, Rodeghier M, Macklin EA, Strunk RC, DeBaun MR. Low forced expiratory volume is associated with earlier death in sickle cell anaemia. Blood. 2015;126(13):1544–50.
36. Sharma S, Efird JT, Knupp C, Kadali R, Liles D, Shiue K, et al. Sleep disorders in adult sickle cell patients. J Clin Sleep Med. 2015;11(3):219–23.
37. Rosen CL, Debaun MR, Strunk RC, Redline S, Seicean S, Craven DI, et al. Obstructive sleep apnea and sickle cell anaemia. Paediatrics. 2014;134(2):273–81.
38. Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1).
39. Chaturvedi S, Labib Ghafuri D, Kassim A, Rodeghier M, DeBaun MR. Elevated tricuspid regurgitant jet velocity, reduced forced expiratory volume in 1 second, and mortality in adults with sickle cell disease. Am J Hematol. 2017;92(2):125–30.
40. Parent F, Bachir D, Inamo J, Lionnet F, Driss F, Loko G, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med. 2011;365(1):44–53.
41. Machado RF, Anthi A, Steinberg MH, Bonds D, Sachdev V, Kato GJ, et al. N-terminal pro-brain natriuretic peptide levels and risk of death in sickle cell disease. JAMA. 2006;296(3):310–8.
42. Mondal P, Yirinec A, Midya V, Sankoorikal BJ, Smink G, Khokhar A, et al. Diagnostic value of spirometry vs impulse oscillometry: a comparative study in children with sickle cell disease. Pediatr Pulmonol. 2019; 54(9):1422–30.
43. Field JJ, Stocks J, Kirkham FJ, Rosen CL, Dietzen DJ, Semon T, et al. Airway hyperresponsiveness in children with sickle cell anaemia. Chest. 2011;139(3): 563–8.
44. Dlamini N, Saunders DE, Bynevelt M, Trompeter S, Cox TC, Bucks RS, et al. Nocturnal oxyhemoglobin desaturation and arteriopathy in a pediatric sickle cell disease cohort. Neurology. 2017;89(24): 2406–12.
45. Barst RJ, Mubarak KK, Machado RF, Ataga KI, Benza RL, Castro O, et al. Exercise capacity and haemodynamics in patients with sickle cell disease with pulmonary hypertension treated with bosentan: results of the ASSET studies. Br J Haematol. 2010;149(3):426–35.
46. Machado RF, Barst RJ, Yovetich NA, Hassell KL, Kato GJ, Gordeuk VR, et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood. 2011;118(4):855–64.
47. Turpin M, Chantalat-Auger C, Parent F, Driss F, Lionnet F, Habibi A, et al. Chronic blood exchange transfusions in the management of pre-capillary pulmonary hypertension complicating sickle cell disease. European Respiratory Journal. 2018;52(4).
48. Knight-Madden JM, Barton-Gooden A, Weaver SR, Reid M, Greenough A. Mortality, asthma, smoking and acute chest syndrome in young adults with sickle cell disease. Lung. 2013;191(1):95–100.
49. Strunk RC, Cohen RT, Cooper BP, Rodeghier M, Kirkham FJ, Warner JO, et al. Wheezing symptoms and parental asthma are associated with a physician diagnosis of asthma in children with sickle cell anaemia. J Pediatr. 2014;164(4):821-6e1.
50. Takahashi T, Okubo Y, Handa A. Acute chest syndrome among children hospitalized with the vaso-occlusive crisis: a nationwide study in the United States. Pediatr Blood Cancer. 2018;65(3):e26885.
51. Glassberg J, Minnitti C, Cromwell C, Cytryn L, Kraus T, Skloot GS, et al. Inhaled steroids reduce pain and sVCAM levels in individuals with sickle cell disease: a triple-blind, randomized trial. Am J Hematol. 2017;92(7):622–31.
52. Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST, Embury S, Moohr JW, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288–94.
53. Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Vasile M, Kasbi F, et al. Chronic and acute anaemia and extracranial internal carotid stenosis are risk factors for silent cerebral infarcts in sickle cell anemia. Blood. 2015;125(10):1653–61.
54. Kassim AA, Pruthi S, Day M, Rodeghier M, Gindville MC, Brodsky MA, et al. Silent cerebral infarcts and cerebral aneurysms are prevalent in adults with sickle cell anemia. Blood. 2016;127(16):2038–40.
55. Powars D, Wilson B, Imbus C, Pegelow C, Allen J. The natural history of stroke in sickle cell disease. Am J Med. 1978;65 (3):461–71.
56. Switzer JA, Hess DC, Nichols FT, Adams RJ. Pathophysiology and treatment of stroke in sickle-cell disease: present and future. Lancet Neurol. 2006;5(6):501–12.
57. DeBaun MR, Gordon M, McKinstry RC, Noetzel MJ, White DA, Sarnaik SA, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N Engl J Med. 2014;371(8):699–710.
58. DeBaun MR, Jordan LC, King AA, Schatz J, Vichinsky E, Fox CK, et al. American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv. 2020; 4(8):1554–88.
59. Dobson SR, Holden KR, Nietert PJ, Cure JK, Laver JH, Disco D, et al. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood. 2002; 99(9):3144–50.
60. Adams RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, et al. Prevention of a first stroke by transfusions in children with sickle cell anaemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339(1):5–11.
61. Vichinsky EP, Neumayr LD, Gold JI, Weiner MW, Rule RR, Truran D, et al. Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anaemia. JAMA. 2010;303(18):1823–31.
62. Hijmans CT, Fijnvandraat K, Grootenhuis MA, van Geloven N, Heijboer H, Peters M, et al. Neurocognitive deficits in children with sickle cell disease: a comprehensive profile. Pediatr Blood Cancer. 2011;56(5): 783–8.
63. Ware RE, Davis BR, Schultz WH, Brown RC, Aygun B, Sarnaik S, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anaemia-TCD With Transfusions Changing to Hydroxyurea (TWiTCH): a multicentre, open-label, phase 3, non-inferiority trial. Lancet. 2016;387(10019):661–70.
64. Scothorn DJ, Price C, Schwartz D, Terrill C, Buchanan GR, Shurney W, et al. Risk of recurrent stroke in children with sickle cell disease receiving blood transfusion therapy for at least five years after the initial stroke. J Pediatr. 2002;140(3):348–54.
65. Hulbert ML, Scothorn DJ, Panepinto JA, Scott JP, Buchanan GR, Sarnaik S, et al. Exchange blood transfusion compared with simple transfusion for first overt stroke is associated with a lower risk of subsequent stroke: a retrospective cohort study of 137 children with sickle cell anaemia. J Pediatr. 2006;149(5):710–2.
66. Hall EM, Leonard J, Smith JL, Guilliams KP, Binkley M, Fallon RJ, et al. Reduction in overt and silent stroke recurrence rate following cerebral revascularization surgery in children with sickle cell disease and severe cerebral vasculopathy. Pediatr Blood Cancer. 2016;63(8):1431–7.
67. Lebensburger JD, Aban I, Pernell B, Kasztan M, Feig DI, Hilliard LM, et al. Hyperfiltration during early childhood precedes albuminuria in pediatric sickle cell nephropathy. Am J Hematol. 2019;94(4):417–23.
68. Niss O, Lane A, Asnani MR, Yee ME, Raj A, Creary S, et al. Progression of albuminuria in patients with sickle cell anemia: a multicenter, longitudinal study. Blood Adv. 2020;4(7):1501–11.
69. Cazenave M, Audard V, Bertocchio JP, Habibi A, Baron S, Prot-Bertoye C, et al. Tubular acidification defect in adults with sickle cell disease. Clin J Am Soc Nephrol. 2020;15(1):16–24.
70. Yee MEM, Lane PA, Archer DR, Joiner CH, Eckman JR, Guasch A. Estimation of glomerular filtration rate using serum cystatin C and creatinine in adults with sickle cell anaemia. Am J Hematol. 2017;92(10): E598–9.
71. Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033–48.
72. Saraf SL, Zhang X, Shah B, Kanias T, Gudehithlu KP, Kittles R, et al. Genetic variants and cell-free hemoglobin processing in sickle cell nephropathy. Haematologica. 2015;100(10):1275–84.
73. Hamideh D, Raj V, Harrington T, Li H, Margolles E, Amole F, et al. Albuminuria correlates with hemolysis and NAG and KIM-1 in patients with sickle cell anaemia. Pediatr Nephrol. 2014;29(10):1997–2003.
74. dos Santos TE, Goncalves RP, Barbosa MC, da Silva GB, Jr., Daher Ede F. Monocyte chemoattractant protein-1: a potential biomarker of the renal lesion and its relation with oxidative status in sickle cell disease. Blood Cells Mol Dis. 2015;54(3):297–301.
75. Audard V, Moutereau S, Vandemelebrouck G, Habibi A, Khellaf M, Grimbert P, et al. First evidence of subclinical renal tubular injury during the sickle-cell crisis. Orphanet J Rare Dis. 2014;9:67.
76. Baddam S, Aban I, Hilliard L, Howard T, Askenazi D, Lebensburger JD. Acute kidney injury during a pediatric sickle cell vaso-occlusive pain crisis. Pediatr Nephrol. 2017;32(8):1451–6.
77. Zahr RS, Hankins JS, Kang G, Li C, Wang WC, Lebensburger J, et al. Hydroxyurea prevents onset and progression of albuminuria in children with sickle cell anaemia. Am J Hematol. 2019;94(1): E27–9.
78. Alvarez O, Miller ST, Wang WC, Luo Z, McCarville MB, Schwartz GJ, et al. Effect of hydroxyurea treatment on renal function parameters: Results from the multi-centre placebo-controlled BABY HUG clinical trial for infants with sickle cell anaemia. Pediatr Blood Cancer. 2012;59(4):668–74.
79. Laurin LP, Nachman PH, Desai PC, Ataga KI, Derebail VK. Hydroxyurea is associated with a lower prevalence of albuminuria in adults with sickle cell disease. Nephrol Dial Transplant. 2014;29(6):1211–8.
80. Quinn CT, Saraf SL, Gordeuk VR, Fitzhugh CD, Creary SE, Bodas P, et al. Losartan for the nephropathy of sickle cell anaemia: a phase-2, multicenter trial. Am J Hematol. 2017;92(9): E520–8.
81. McClellan AC, Luthi JC, Lynch JR, Soucie JM, Kulkarni R, Guasch A, et al. High one-year mortality in adults with sickle cell disease and end-stage renal disease. Br J Haematol. 2012;159(3):360–7.
82. Gérardin C, Moktefi A, Couchoud C, Duquesne A, Ouali N, Gataut P, et al. Survival and specific outcome of sickle cell disease patients after renal transplantation. Br J Haematol. 2019;187(5):676–80.
83. Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376(5):429 –39.
84. Niihara Y, Miller ST, Kanter J, Lanzkron S, Smith WR, Hsu LL, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. 2018;379(3):226–35.
85. Vichinsky E, Hoppe CC, Ataga KI, Ware RE, Nduba V, El-Beshlawy A, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 2019;381(6): 509–19.
86. Gluckman E, Cappelli B, Bernaudin F, Labopin M, Volt F, Carreras J, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2017; 129 (11):1548–56.
87. Eapen M, Brazauskas R, Walters MC, Bernaudin F, Bo-Subait K, Fitzhugh CD, et al. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: a retrospective multicentre, cohort study. Lancet Haematol. 2019;6(11):e585–96.
88. Bolanos-Meade J, Cooke KR, Gamper CJ, Ali SA, Ambinder RF, Borrello IM, et al. Effect of increased dose of total body irradiation on graft failure associated with HLA-haploidentical transplantation in patients with severe haemoglobinopathies: a prospective clinical trial. Lancet Haematol. 2019;6(4) :e183–93.
89. Krishnamurti L, Ross D, Sinha C, Leong T, Bakshi N, Mittal N, et al. Comparative effectiveness of a web-based patient decision aid for therapeutic options for sickle cell disease: a randomized controlled trial. J Med Internet Res. 2019;21(12):e 14462.
Credits: Brandow, A.M., Liem, R.I. Advances in the diagnosis and treatment of sickle cell disease. J Hematol Oncol 15, 20 (2022). https://doi.org/10.1186/ s13045-022-01237-z






